Hematology Glossary
A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | X | Y | Z
ABack to top ^ | ||
| Abciximab | A monoclonal antibody (immunoglobulin) that binds glycoprotein IIb/IIIa on platelets and inhibits platelet function/aggregation. This particular drug has been used in percutaneous transluminal coronary artery stenting procedures. Note: Drugs ending with "mab" are "Monoclonal AntiBodies". | |
| ABO blood groups | Specific antigen system located on red blood cells (RBCs) and platelets (e.g. blood type A, B, AB or O). ABO blood type determination is an essential part of pre-transfusion testing for patients. (Note: Patients with type A RBCs will have type B antibody in their plasma, while patients with type B RBCs will have type A antibody in their plasma. Since type O patient's red blood cells lack A antigen and B antigen, then not surprisingly they could have antibodies to both A and B antigens in their plasma. On the other hand, AB patients plasma is typically devoid of antibodies to either A or B antigen. Hence, the universal donor for RBCs is type O (devoid of A or B antigen) and universal donor for plasma is type AB (devoid of anti-A or Anti-B anti antibodies). | |
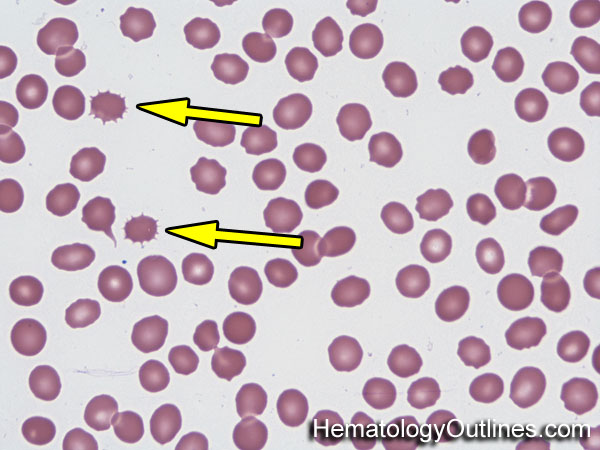
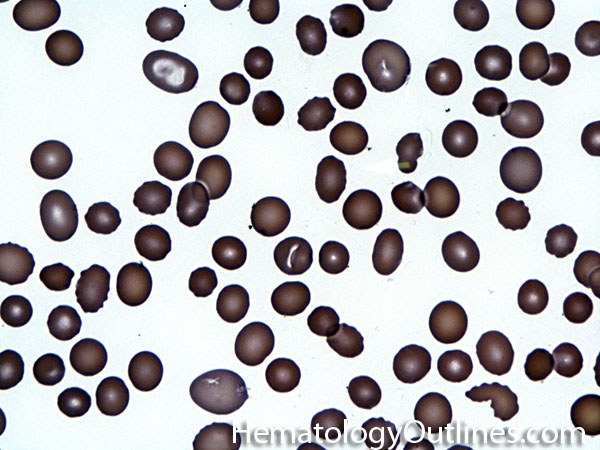
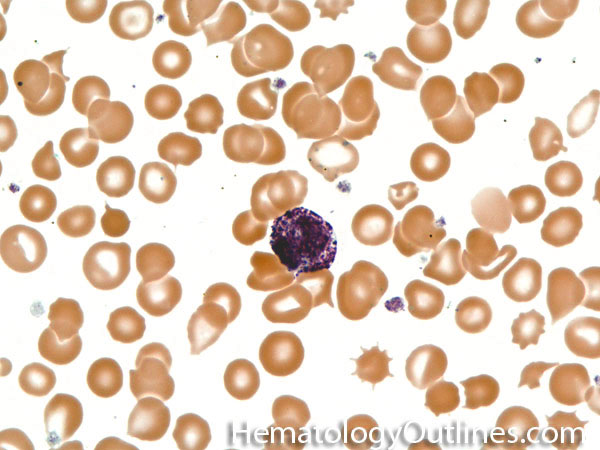
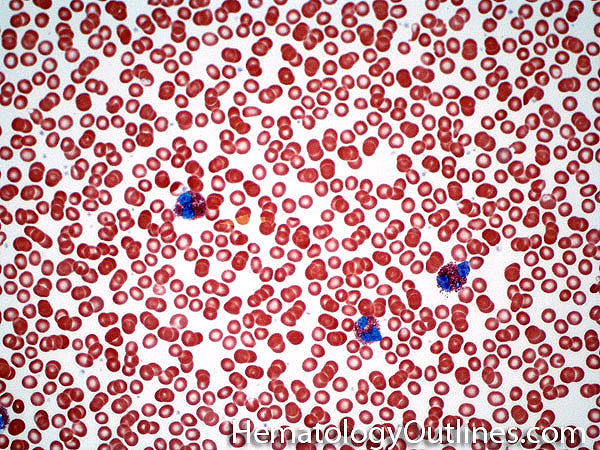
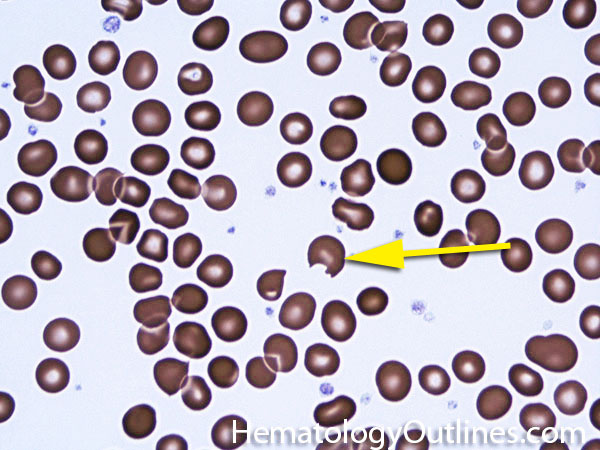
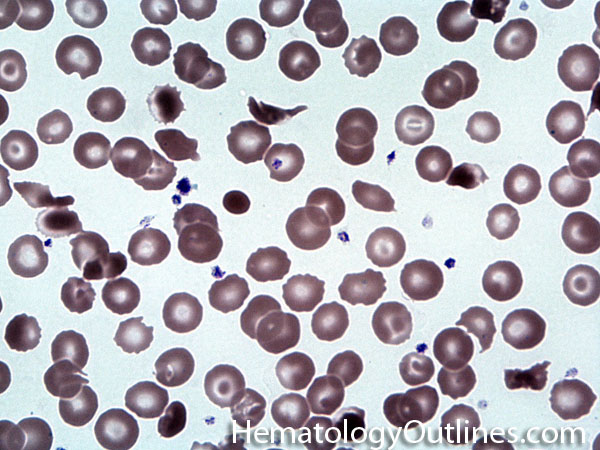
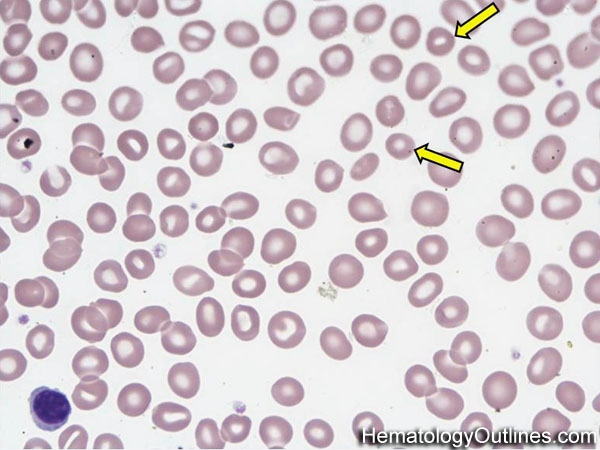
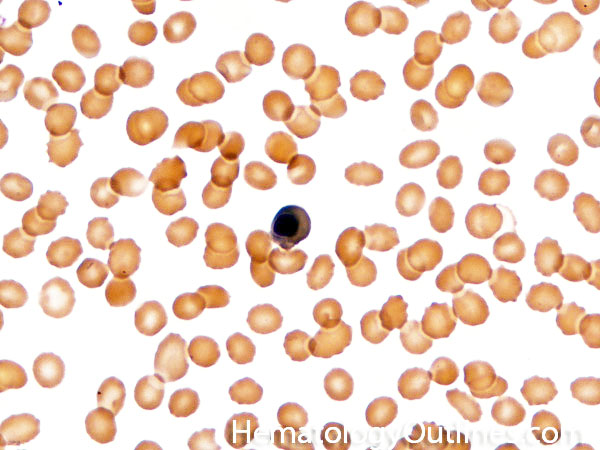
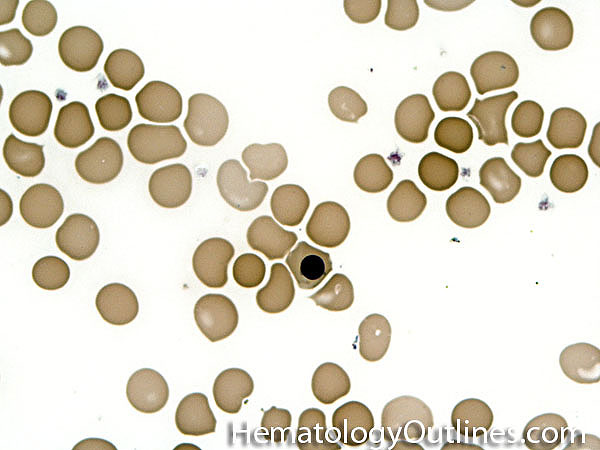
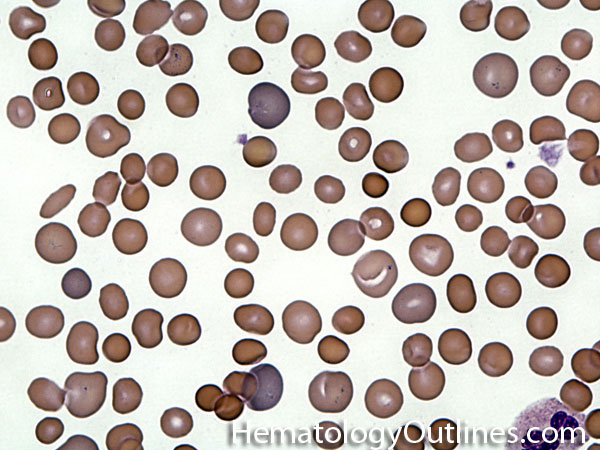
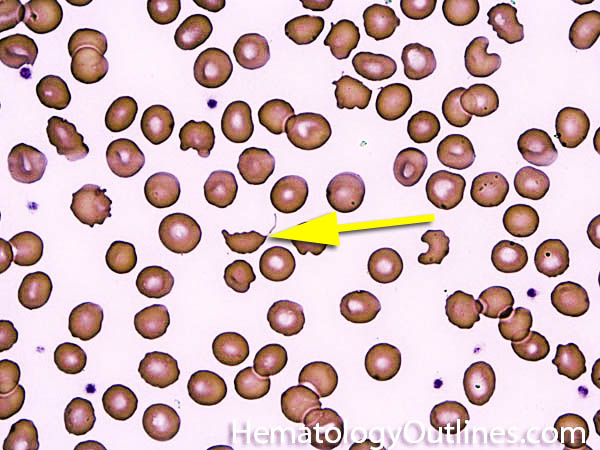
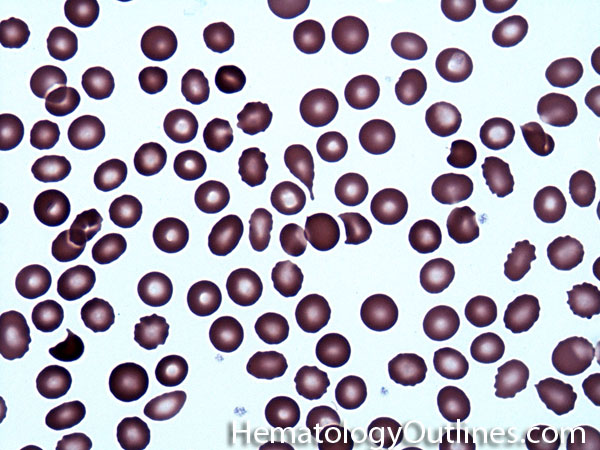
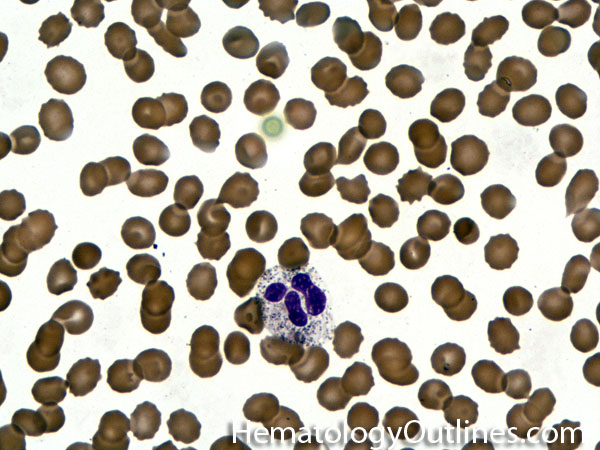
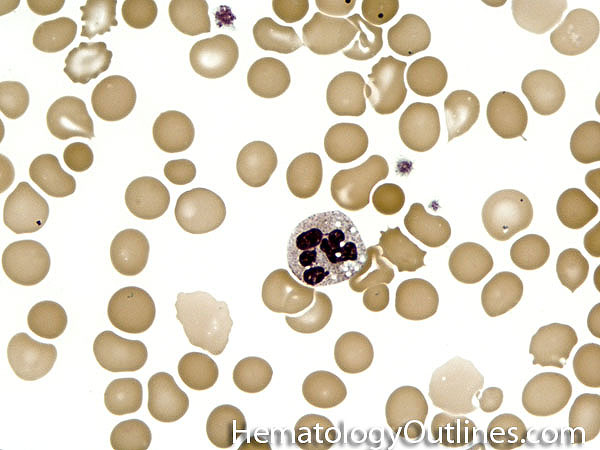
| Acanthocyte | Also known as Spur Cell is an RBC with irregular thorn-like projections. Looks like "Cowboy Boot Spurs". These RBCs may be seen in some peripheral smears due to artifacts or may be associated with certain conditions including Abetalipoproteinemia, Liver disease, Malnutrition, Hypothyroidism, Asplenia, and McLeod Phenotype of Kell blood group, etc.
  |
|
| Acetylsalicylic acid | a.k.a. aspirin belongs to the non-steroid anti-inflammatory drug (NSAID) family which is used as an analgesic, antipyretic, and anti-inflammatory agent. Because of its anti-platelet properties, it is used in low dosage to prevent myocardial infarction, stroke, and thrombus formation. Main mechanism of actions includes irreversible binding of cyclooxygenase-1 (COX-1) and modification of Cox-2, thus decreases the production of prostaglandin and thromboxane A2. Hence, irreversibly effects the function of the platelets and may be problematic if the patient is on Aspirin and planned for a surgical procedure. Additionally, usage in children or adolescents for fever or other illnesses can cause Reye's syndrome. | |
| Activated Partial Thromboplastin Time | Also known as aPTT or PTT, is laboratory test that measures the integrity of the intrinsic pathway of coagulation and commonly used for monitoring heparin therapy. It is sensitive to detecting deficiencies in factors involved in the intrinsic pathway such as factor VIII, IX, XI, and XII common pathway such as factor X or phospholipid-dependent inhibitors such as lupus anticoagulants. As noted above, it is also prolonged if certain anticoagulants such as heparin are present. The test is performed by taking citrated plasma to clot at 37 degrees C after the addition of calcium (a set amount of phospholipid is also added but no tissue factor is added so it will not evaluate the extrinsic pathway). PTT could also be used comparatively in mixing studies when one suspects factor deficiency or presence of inhibitors. In a patient with prolonged PTT (& normal PT), first the presence of heparin must be excluded. Once heparin is excluded, the patient's plasma can be mixed with a known normal plasma sample to see if the PTT corrects. Correction of the PTT in such setting is suggestive of a factor deficiency while lack of correction may imply presence of an inhibitor (antibody), such as lupus anticoagulant. | |
| Activated-protein C resistance | APC resistance can be either acquired or inherited. The most common cause of APC resistance is due to a mutation at the cleavage site in Factor V, in which the amino acid Arg506 is replaced with Gln, producing Factor V Leiden. This mutation prevents APC from inactivating Factor V. APC resistance is usually associated with Factor V Leiden and results in an increased risk of thrombosis. | |
| Acute | Generally refers to a process or disease that starts abruptly and/or progresses quickly. Examples include acute lymphoblastic Leukemia (AKA ALL or Lymphoblastic leukemia/Lymphoma) & acute myeloid leukemia (AML) for neoplastic processes. | |
| Acute lymphoblastic leukemia | Also known as ALL is an Acute Leukemia of lymphoid origin (B-cell origin is called B-ALL and T-cell origin is called T-ALL). Usually the peripheral blood, tissue and/or bone marrow shows an increase in number of lymphoblasts (typically greater than 20% in the peripheral blood &/or bone marrow as seen in many cases of B-ALL).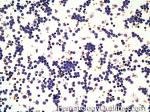 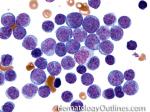 |
|
| Acute myeloid leukemia | Acute Leukemia of myeloid origin. Peripheral blood, tissue and/or bone marrow show increased number of myeloblasts (typically greater than 20% in the peripheral blood &/or bone marrow). Based on WHO-2008 criteria AMLs are divided into 1) AMLs with recurrent cytogenetic abnormalities (which typically have a better prognosis than the other AMLs) such as APL with t(15;17), AML with t(8;21), AML with t(16;16) or inv (16). 2) AML with Myelodysplasia-related changes (usually have a poor prognosis). 3) Therapy-Related AMLs (usually have a poor prognosis) and 4) AML-NOS which can be minimally differentiated, granulocytic origin (e.g. AML-M2), monocytic or myelomonocytic origin (AML-M4 & M5), erythroid origin (AML-M6) , or megakaryocytic origin (AML-M7). The prognosis on AML-NOS group is variable. Note: A blast with Auer rod(s) is by definition a myeloblast. Myeloblasts with auer rod(s) have been noted in some AML-NOS (e.g. M2) and many times noted in APL. | |
| Acute phase response | Is also known as Acute phase reaction and is an inflammatory response to injury which may be related to an underlying infection, trauma, ischemia, immunologic or neoplastic process. During this reaction, the liver produces "acute phase proteins" which include but are not limited to CRP, fibrinogen, Factor VIII, ferritin, hepcidin, Ceruloplasmin and haptoglobin. Hence, evaluating the levels of these proteins in the setting of inflammation may be problematic. | |
| ADAMTS-13 | A member of the metalloprotease family which is responsible for cleaving high molecular weight von Willebrand Factors (vWF). The cleavage of the vWF multimers reduces their size. Note: In Thrombotic Thrombocytopenic Purpura (TTP) there is a decrease or absence of ADAMTS-13 which leads to larger vWF multimers that increases the chance of forming platelet thrombi and leading to microvascular thrombosis. | |
| Adult T-cell Leukemia/Lymphoma | A systemic mature T-cell neoplasm that is associated with HTLV1 virus infection. Hence, this Leukemia/Lymphoma is more common in certain endemic areas such as parts of Japan, Africa and Caribbean islands. | |
| Adverse hematologic reactions secondary to drugs | Adverse hematologic reactions to drugs include but are not limited to bone marrow suppression, agranulocytosis, thrombocytopenia, megaloblastoid changes, etc. | |
| Afibrinogenemia | Afibrinogenemia and hypofibrinogenemia, refers to absence or reduction in the amount of the clotting factor fibrinogen in the blood. This is seen in rare instances as an inherited disorder, but more commonly it is seen with disseminated intravascular coagulation (DIC). | |
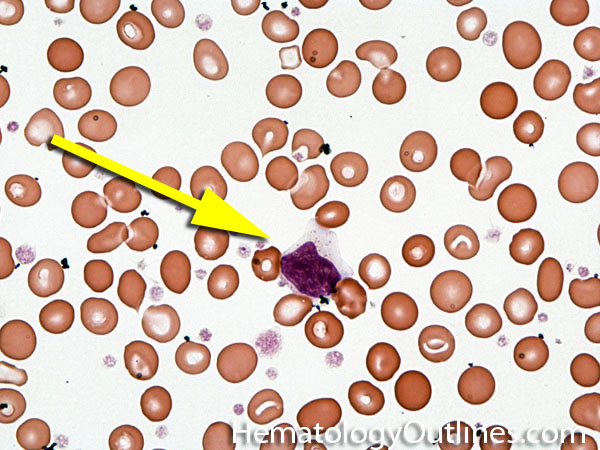
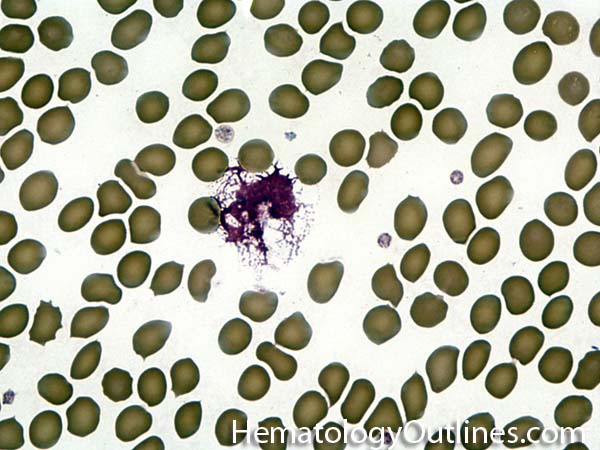
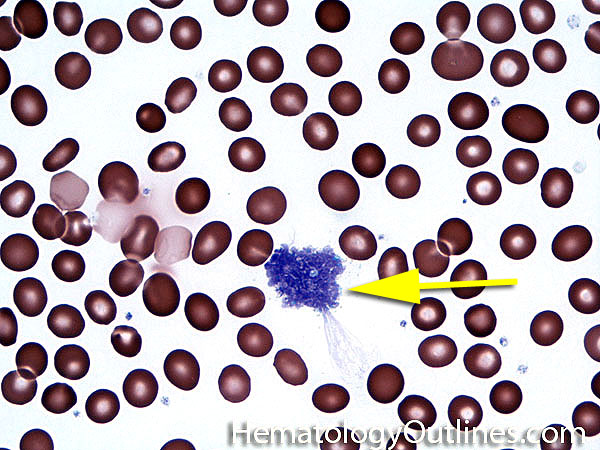
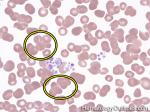
| Agglutination | Aggregation or clumping of biologic substances such as bacteria or cells (e.g. red blood cell agglutination as noted in direct and indirect antiglobulin test, AKA Coombs' test). Agglutination may also be indicative of antibody-antigen presence on coated cells or substances.
 |
|
| Agranulocytosis | An adverse reaction that is usually due to certain drugs that leads to severe neutropenia and may be followed by fever and sore throat. Untreated, it may be deadly. Many drugs can cause agranulocytosis including but not limited to certain antibiotics, anti-thyroid medications, antihypertensive drugs, antipsychotic drugs, anti-inflammatory drugs, anticonvulsants, hypoglycemic drugs, and antidepressants. | |
| AIHA | AutoImmune Hemolytic anemia is usually due to autoantibodies (usually IgG) directed against RBCs. Hemolyic anemia results when splenic macrophages recognize the antibodies on the red cells resulting in phagocytosis and destruction. Some red cells escape complete destruction but loose membrane and surface area, resulting in circulating spherocytes. | |
| Albumin | A major protein constituent of blood plasma. This protein is relatively negatively charged (as opposed to immunoglobulins which are relatively positively charged) and serves as a major protein in the plasma. It is produced in the liver so levels are usually reduced in end stage liver disease. | |
| ALCL | Also known as anaplastic large cell Lymphoma is usually an aggressive mature/maturing T-cell lymphoma that typically involve nodal tissue (e.g. lymph nodes) and sometimes skin (Cutaneous ALCL has better prognosis). The nodal ALCLs can be either ALK (Anaplastic Large cell Kinase) positive (better prognosis and a potential source of therapy) or ALK negative (worse prognosis in nodal ALCLs). Please note that the cutaneous ALCL is typically ALK negative with best prognosis. In Summary: Amongst ALCLs, ALK negative cutaneous ALCL has the best prognosis while the Nodal ALK negative ALCL has the worst prognosis. | |
| Alder-Reilly Anomaly | Evenly distributed violet purple granules without Dohle bodies or vacuoles in the cytoplasm of all neutrophils. Looks like toxic granules but unlike toxic granules, the findings are present in all the neutrophils and can also be seen in monocytes and lymphocytes. Please note that the granulocytes appear to be functionally normal. Hence, it's called an anomaly and not a disease. | |
| Alemtuzumab | Also known as CAMPATH is a Monoclonal antibody used in treating certain hematologic malignancies. Its mechanism of action is based on binding CD52 on cells and resulting in lysis. Note: Drugs ending with "MAB" are "Monoclonal AntiBodies". | |
| Alkylating agents | A family of cytotoxic agents that are used in treatment of certain hematologic neoplasms. Their mechanism of action involves altering DNA and interfering with replication and transcription. Patients treated with these agents are at long term risk of developing other post treatment neoplastic disorders (e.g. Myeloid disorders such as MDS and AML). | |
| All-Trans Retinoic Acid | a.k.a. ATRA is an acid form of vitamin A, used to treat Acute Promyelocytic Leukemia (APL). | |
| Alloantibody | These are antibodies that recognizes non-self antigens. Example: Alloantibodies can develop in patients who have received prior transfusions. Hence, having formed antibodies to non-self red blood cell antigens (e.g. Anti-Kell, etc.). If a patient develops an alloantibody to Kell positive RBC, the next time this patient receives a Kell positive RBC transfusion, he or she may adversely react to the transfused blood and develop a hemolytic transfusion reaction which could be deadly. This is a major reason for testing patient's blood prior to transfusing them with blood products. | |
| Allogeneic | Typically refers to transferring tissue (e.g. bone marrow), blood, stem cells from one individual to another individual (e.g. allogeneic stem cell transplantation). The opposite of autologous which refers to being from self. | |
| Alloimmunization | Formation of antibodies to non-self cellular antigens. Example includes formation of antibodies to non-self red blood cell antigens (e.g. Anti-Kell, etc.). | |
| Allopurinol | Brand name: Zyloprim is a purine analog drug used to treat hyperuricemia such as in the setting of gout. Rare but potentially fatal side effects include Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN). Can cause severe pancytopenia when used with azathioprine or mercaptopurine. | |
| Allotype | It is usually a resultant of qualitative or quantitative difference in blood cell surface antigens between different individuals due to genetic polymorphism. Ultimately leading to antigenic variation which can be recognized by alloantibodies. | |
| Alpha granules | A type pf platelet granule.There are three types of granules present within platelets: Alpha granules, Delta (dense) granules and Lysosomes. Alpha granules contain the larger molecules such as Platelet Factor 4 (PF4), Platelet-derived growth factor (PDGF), fibrinogen and some other clotting factors. The Delta (Dense) Granules contain the smaller molecules such as Serotonin, ADP, & Calcium. Mnemonic is Alpha stands for the "Bigger" molecules such as proteins and peptides, while Delta or Dense stands for "Diminutive" (tiny) molecules. | |
| Alpha Storage Pool Disease | AKA gray platelet syndrome is characterized by thrombocytopenia and abnormal enlarged gray-blue platelets with a washed-out appearance due to reduced alpha granules in platelets. | |
| Alpha thalassemia | A type of thalassemia (Red blood cell disorder) that is specifically due to underproduction of the Alpha hemoglobin chain. As opposed to beta thalassemia which is due to underproduction of Beta hemoglobin chain. Note: As opposed to hemoglobinopathies which have intrinsic defects in the hemoglobin chain leading to abnormal hemoglobin molecules (e.g. sickle cell anemia), in thalassemias the hemoglobin molecule is usually not abnormal but rather underproduced. | |
| Alpha-2-antiplasmin | A serine protease inhibitor that inactivates plasmin and fibrinolysis. | |
| AML | Acute Myeloid Leukemia (AML) is an acute leukemia of myeloid origin. Peripheral blood, tissue and/or bone marrow show increased number of myeloblasts (typically greater than 20% in the peripheral blood &/or bone marrow). Based on WHO-2008 criteria AMLs are divided into 1) AMLs with recurrent cytogenetic abnormalities (which typically have a better prognosis than the other AMLs) such as APL with t(15;17), AML with t(8;21), AML with t(16;16) or inv (16). 2) AML with Myelodysplasia-related changes (usually have a poor prognosis). 3) Therapy-Related AMLs (usually have a poor prognosis) and 4) AML-NOS which can be minimally differentiated, granulocytic origin (e.g. AML-M2), monocytic or myelomonocytic origin (AML-M4 & M5), erythroid origin (AML-M6) , or megakaryocytic origin (AML-M7). The prognosis on AML-NOS group is variable. Note: A blast with Auer rod(s) is by definition a myeloblast. Myeloblasts with auer rod(s) have been noted in some AML-NOS (e.g. M2) and many times noted in APL. | |
| Amyloidosis | A disease associated with deposition of amyloid in tissues. There are multiple different types of amyloids but the one typically seen in hematologic disorders is the AL (Amyloid Light chain) type which usually is associated with an underlying plasma cell neoplasm. The AL amyloid is associated with plasma cell neoplasms and are usually monoclonal light chains (kappa or lambda) leading to linear nonbranching fibril aggregates measuring 7.5-10 nm in width and ultimately comprised of beta-pleated sheets which can be highlighted with the congo red stain in tissue. | |
| Anaplastic large cell lymphoma | Also known as ALCL is usually an aggressive mature/maturing T-cell Lymphoma that typically involve nodal tissue (e.g. lymph nodes) and sometimes skin (Cutaneous ALCL has better prognosis). The nodal ALCLs can be either ALK (Anaplastic Large cell Kinase) positive (better prognosis and a potential source of therapy) or ALK negative (worse prognosis in nodal ALCLs). Please note that the cutaneous ALCL is typically ALK negative with best prognosis. In Summary: Amongst ALCLs, ALK negative cutaneous ALCL has the best prognosis while the Nodal ALK negative ALCL has the worst prognosis. | |
| Anemia | Refers to a decrease in hematocrit/hemoglobin levels (e.g. iron deficiency anemia). Anemias are typically classified into 3 broad categories: microcytic Anemia (MCV <80), normocytic Anemia (MCV 80-100) and macrocytic Anemia (MCV >100). | |
| Angioimmunoblastic T-cell lymphoma | These are usually aggressive mature/maturing T-cell lymphomas that typically involve nodal tissue (lymph nodes). Patients with this Lymphoma may have an underlying immune dysfunction and many are secondarily associated with EBV virus. 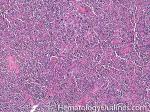 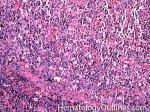 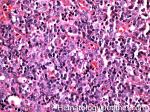 |
|
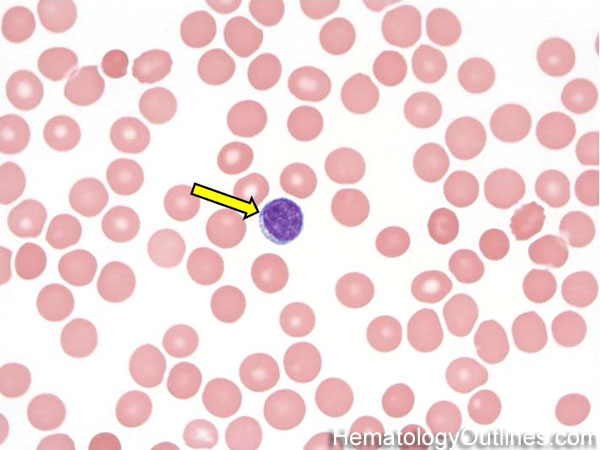
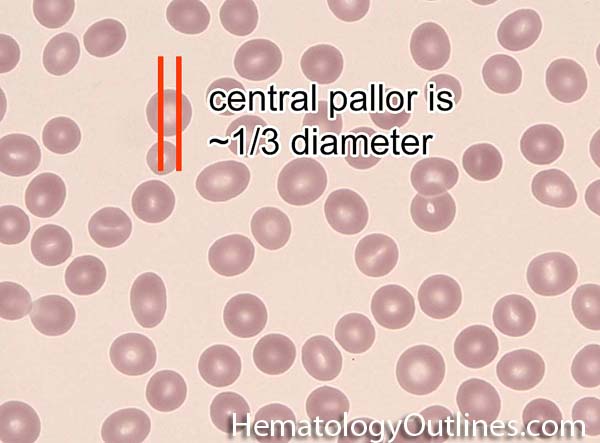
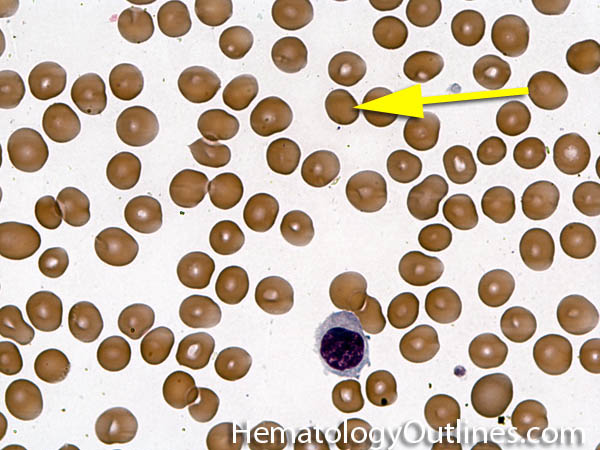
| Anisocytosis | Refers to the variation in "size" of red blood cells. Laboratory value that correlates with anisocytosis is an elevated RDW.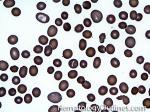 |
|
| Anti-D immunoglobulin | antibody formed to one of the Rh antigens on red blood cells known as D antigen which can lead to red blood cell destruction. Classic example of this is seen in hemolytic disease of the newborn where the mother's anti-D antibodies attack and destroy the fetal Rh (D) positive red blood cells. | |
| Anti-phospholipid syndrome | An autoimmune syndrome characterized by antibodies against phospholipids that causes thrombosis such as anti-cardiolipin antibodies, lupus anticoagulant antibodies, and anti-beta2 glycoprotein I antibodies. It is commonly associated with SLE and other autoimmune diseases. It can cause thrombosis and pregnancy-related complications such as preeclampsia and spontaneous abortion. Testing includes PTT, lupus anticoagulant antibody with dRVVT and testing for anticardiolipin. | |
| Antibody | Also known as immunoglobulin is a protein that is produced by B cells (in terminally differentiated B-cells known as plasma cells these immunoglobulins are produced and secreted and are not cell surface bound) which recognize and bind to other proteins or substances (foreign or self antigens). Antibodies can be part of an immune response and help eradicate viruses and bacteria or may be part of an autoimmune destructive processes in hematology such as autoimmune hemolytic anemia or immune mediated platelet destruction (e.g. ITP). | |
| Anticoagulant | These are natural or synthetic compounds that prevent the formation of blood clots. Common examples include heparin, warfarin (coumadin), etc. | |
| Antigen | Typically refers to a molecular structure (on a foreign substance or self) which can be recognized by an antibody (e.g. foreign antigens include viral and bacterial components while self-antigens include certain aspects of a platelet or red blood cell, etc.). | |
| APC resistance | a.k.a. activated-Protein C resistance can be either acquired or inherited. The most common cause of APC resistance is due to a mutation at the cleavage site in Factor V, in which the amino acid Arg506 is replaced with Gln, producing Factor V Leiden. This mutation prevents APC from inactivating Factor V. APC resistance is usually associated with Factor V Leiden and results in an increased risk of thrombosis. | |
| APL | Acute Promyelocytic Leukemia. Usually has the following translocation, t(15;17) and is responsive to ATRA therapy.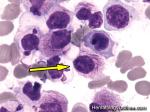 |
|
| Aplastic anemia | A rare disorder that results in underproduction of bone marrow hematopoietic cells. Hence patients usually present with pancytopenia and their bone marrow appears markedly hypocellular (usually <10% cellularity). Aplastic anemia can be secondary to multiple causes including but not limited to drugs, infections (e.g. viruses such as EBV and parvovirus), neoplastic disorders (e.g. myelodysplastic syndrome or paroxysmal nocturnal hemoglobinuria), and autoimmune disorders. | |
| Apoptosis | A form of cell death. As opposed to necrosis which is typically an abnormal process, apoptosis can be secondary to both normal and abnormal processes. Normal apoptosis is seen in lymph node germinal centers where the normal B-cells that are not selected undergo apoptosis. Abnormal processes with apoptosis include high grade lymphomas such as Burkitt_lymphoma where there is a high rate of mitosis which leads to increased number of cells undergoing apoptosis.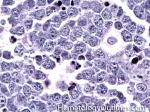 |
|
| Argatroban | Belongs to the class of univalent direct thrombin inhibitors, which also include Melagatran and Dabigatran. Called univalent because they bind only to the active site of the thrombin molecule. Used to treat heparin-Induced Thrombocytopenia and can be monitored through PTT. It is cleared by the liver and therefore can be used in patients with renal insufficiencies. | |
| Aspirin | a.k.a. acetylsalicylic acid belongs to the non-steroid anti-inflammatory drug (NSAID) family which is used as an analgesic, antipyretic, and anti-inflammatory agent. Because of its anti-platelet properties, it is used in low dosage to prevent myocardial infarction, stroke, and thrombus formation. Main mechanism of actions includes irreversible binding of cyclooxygenase-1 (COX-1) and modification of Cox-2, thus decreases the production of prostaglandin and thromboxane A2. Hence, irreversibly effects the function of the platelets and may be problematic if the patient is on Aspirin and planned for a surgical procedure. Additionally, usage in children or adolescents for fever or other illnesses can cause Reye’s syndrome. | |
| ATRA | All-Trans Retinoic Acid. Acid form of vitamin A which is used in treatment of Acute Promyelocytic Leukemia (APL). | |
| Auer rods | These are abnormal pink needle shaped polymerized granular structures found in the cytoplasm of abnormal myeloblasts that may be seen in certain myeloid neoplasms. Presence of Auer rods is typically associated with Acute myeloid Leukemia (AML) and rarely associated with Refractory anemia with Excess Blasts II (RAEB-II) which is the most aggressive type of MDS and usually progresses to AML.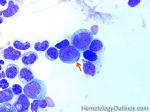 |
|
| Autoantibodies | These are antibodies that recognize self-antigens (e.g. Autoimmune Hemolytic anemia which can have autoantibodies, usually IgG, to RBCs and ultimately have the IgG-coated RBCs destroyed in the spleen, known as extravascular hemolysis). | |
| Autologous | Typically refers to transferring tissue (e.g. bone marrow), blood, or stem cells from ones body to self (e.g. autologous stem cell transplantation). | |
BBack to top ^ | ||
| B-ALL | Also known as B-Lymphoblastic Leukemia/Lymphoma (or B-Acute lymphoblastic leukemia) is an Acute leukemia of lymphoid origin (specifically B-cell origin). Usually the peripheral blood, tissue and/or bone marrow shows an increase in number of lymphoblasts (typically greater than 20% in the peripheral blood &/or bone marrow as seen in many cases of B-ALL). | |
| B-cell | Type of white blood cell and more specifically a type of lymphocyte which produces antibodies (immunoglobulins). B-cells (like other hematopoietic cells) originate in the marrow and start maturation in the bone marrow and complete their maturation process in germinal center of secondary follicles within secondary lymphoid tissues/organs such as lymph nodes, mucosa associated lymphoid tissue (MALT), or spleen. Remember B-cell for Bone marrow and T-cell for Thymus (B-cells start maturation in Bone marrow while T-cells mature in the Thymus.
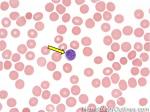 |
|
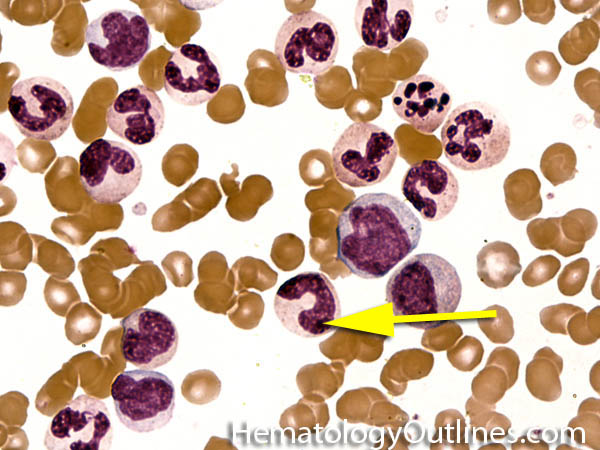
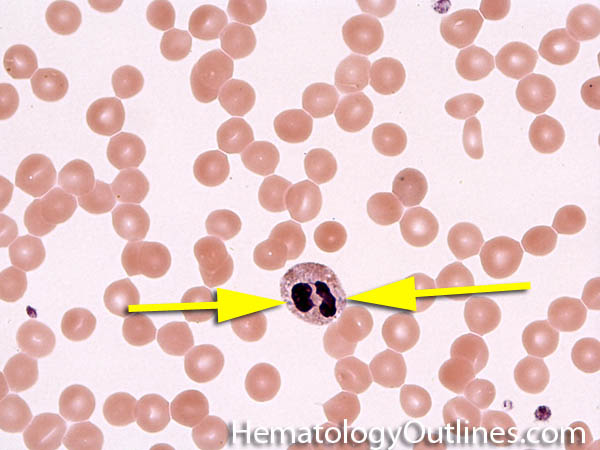
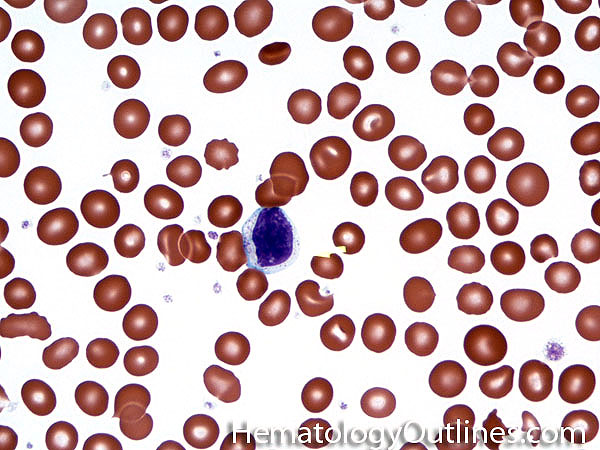
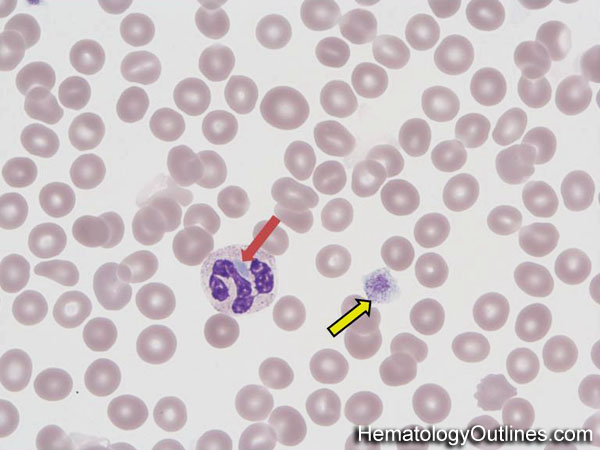
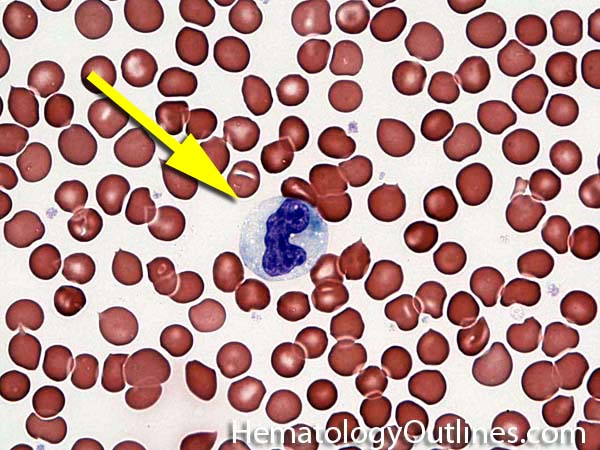
| Band | Refers to Band form of Neutrophil. A maturing neutrophil that precedes the segmented neutrophils (PMNs) in the granulocytic maturation scheme. Increased numbers of Bands may be indicative of an underlying infection (especially bacterial).
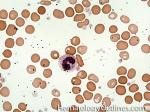   |
|
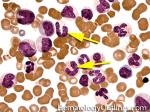
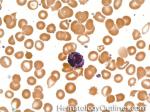
| Basophils | Type of white blood cell and more specifically granulocyte that maybe elevated in certain conditions such as but not limited to allergic reactions or CML.
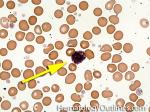  |
|
| Benign | Refers to a lesion or tumor that typically will not metastasize. The opposite of malignant. | |
| Beta thalassemia | Beta thalassemia is due to underproduction of the beta hemoglobin chain. Due to underproduction of this beta chain, other globin chains such as the delta chain will compensate which is manifested by the increased level of Hemoglobin A2 (recall comprised of 2 alpha chains and 2 delta chains) noted in many of these patients. | |
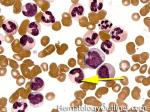
| Blast | Refers to an immature precursor of a white blood cell. Two types are Myeloid (myeloblast) and Lymphoid (lymphoblast).
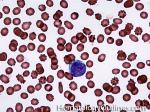 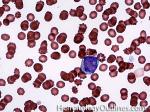 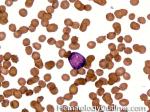 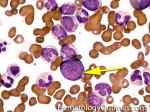 |
|
| Bleeding disorder | A disease that is typically due to clotting problems, either due to a defect or deficiency of platelets, platelet associated factors or clotting factors. Common examples include but are not limited to hemophilia A (Factor VIII deficiency), severe thrombocytopenia (e.g. platelet counts of <10,000) and von Willebrand's disease (deficiency or defect of the vW factor. | |
| Blood | Special fluid that is within vessels and is comprised of the cellular components (e.g. red blood cells, white blood cells and platelets) and plasma with proteins (e.g. clotting factors, vW factor, etc.).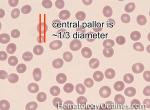 |
|
| Blood Bank | A section in most hospital's laboratory area which is responsible for collection, separation and storage of blood products such as Red Blood Cells, platelets, Fresh Frozen Plasma, etc. | |
| Blood Smear | A glass slide prepared from peripheral blood for microscopic evaluation of the blood elements (e.g. Red Blood Cells, white blood cells, platelets, etc.). | |
| Bone marrow | The soft spongy cellular area in between the trabecular bony areas inside of the bone where blood's cellular components (e.g. Red Blood Cells, white blood cells, and platelets) are produced. | |
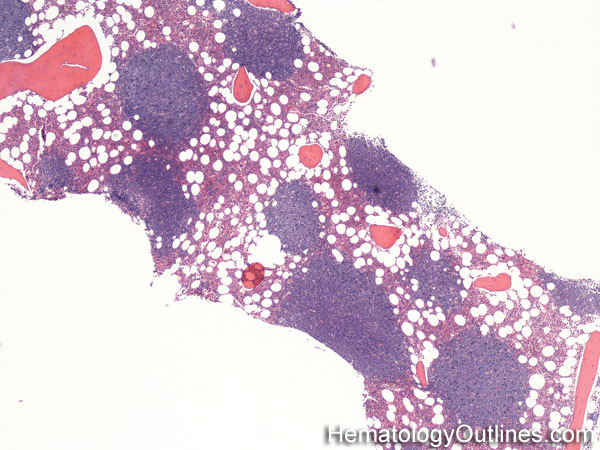
| Bone marrow serous fat atrophy | This usually refers to a hypocellular bone marrow with fat atrophy, and deposition of extracellular "gelatinous-like" material. This may be due to a damaged hematopoietic inductive microenvironment in the bone marrow and may be secondary to therapy (e.g. chemotherapy), HIV, or severe malnutrition. | |
| Bone Marrow Transplant | Transfer of healthy bone marrow cells to an individual with diseased or damaged bone marrow. | |
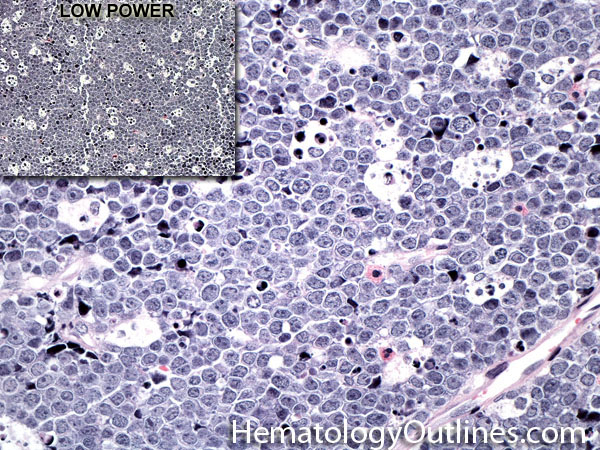
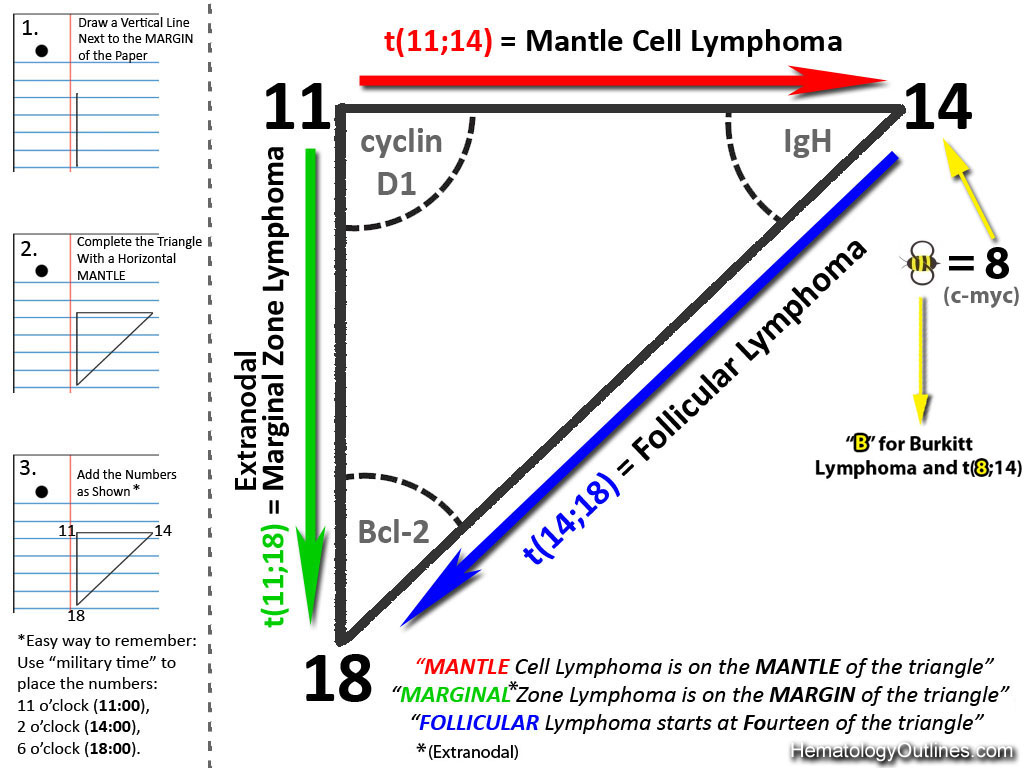
| Burkitt lymphoma | An aggressive mature/maturing B-cell Lymphoma which is characterized by intermediate-sized cells with a very high mitotic index (ki67 close to %100). The typical immunophenotype is CD19+, CD20+, CD10+, CD5-, and BCL2-. The classic translocation associated with this lymphoma is the t(8;14) which involves c-MYC and IgH (see lymphoma mnemonic diagram below for an easy way to remember this and other common B-cell lymphomas).
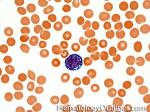 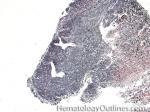   |
|
CBack to top ^ | ||
| Cancer | An abnormal growth of malignant cells with a chance of spreading (metastasis) to other sites. | |
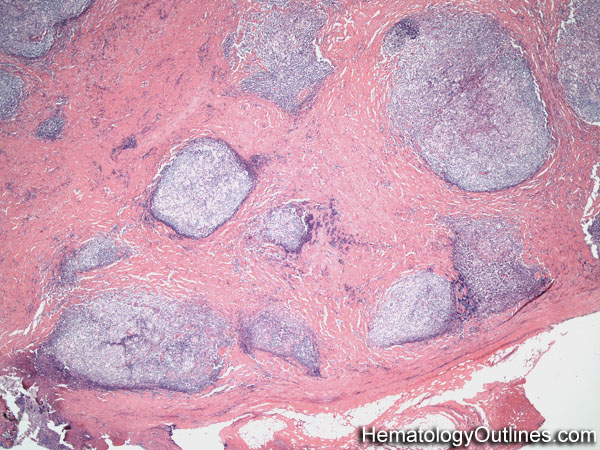
| Castleman disease | A heterogenous group of diseases that present with lymphadenopathy with variable presentations and prognosis. There are 3 main types recognized: 1) Hyaline vascular variant (the most common subtype, more common in younger patients, usually asymptomatic, localized disease, and best prognosis) ; 2) plasma cell variant (Many are localized and more common in elderly with systemic symptoms such as anemia, thrombocytopenia, elevated ESR, elevated IL6, and polyclonal hypergammaglobulinemia); 3) Multicentric variant (Some similarities to plasma cell variant with similar systemic symptoms and more common in elderly. However, this variant as the name implies usually involves multiple lymph nodes, is often seen in HIV positive patients, usually HHV8 positive, and has the worst prognosis. Additionally, it can be also associated with other HHV8 positive neoplasms such Kaposi Sarcoma).  |
|
| CBC | Complete blood Count which includes a count of the WBC, hemoglobin, hematocrit and platelets. When a differential count is added to the CBC, the different types of WBCs are quantitated (e.g. percent neutrophils, lymphocytes, monocytes, eosinophils and basophils). | |
| CD | Stands for "cluster of differentiation" which are usually cell surface glycoproteins expressed by normal and abnormal cells and used primarily in diagnostic hematology as part of the workup of hematologic malignancies by either flow cytometry or immunohistochemistry (e.g. co-expression of CD5 and CD23 by the neoplastic cells in CLL). | |
| CD Mnemonic | The mnemonic is "T" for "Tiny" CDs and "T-cells" referring to the fact that typically the Tiny CDs (CD1, 2, 3, 4, 5, 6, 7, & 8) are expressed in T-cells as opposed to "B" for "B-cells" & "Bigger" CDs (CD19, 20, 21, 22, 23, and 24) which are typically expressed in "B-cells". The "Middle" CDs (CD11, 12, 13, 14, 15, 16, 17 & 18) are more commonly expressed in "MyeloMonocytic" (M for Middle and M for Myelomonocytic referring to granulocytes and monocytes). Note: The above mnemonic is mainly for the normal T-cells, Myeloid cells and B-cells. Abberant expression may be seen in other lineages in diseased states and some common examples include the expression of the T-cell marker CD7 on abnormal myeloblasts in some AMLs or the abnormal expression of the T-cell marker CD5 on CLL or Mantle Cell Lymphoma. | |
| CD1a | A type of CD which is normally expressed in immature thymic T-cells (e.g. cortical thymocytes) and dendritic cells.Recall the Mnemonic for CDs. Also remember that since this is the lowest numbered CD, then it is typically only expressed in immature T-cells (cortical thymocytes) and not present in more mature T-cells. Recall the CD_Mnemonic. | |
| CD2 | A type of CD which is normally expressed in most T-cells (immature and mature). Recall the CD_Mnemonic. | |
| CD3 | A type of CD which is normally expressed in most T-cells (immature and mature). CD3 is the most specific T-cell marker. Recall the CD_Mnemonic. | |
| CD4 | A type of CD which is normally expressed in a subset of T-cells (Helper T-cells). Recall the CD_Mnemonic. This CD is also expressed in lower levels on monocytes. Also recall the rule of 8 mnemonic: CD4 associates with MCH Class II (4x2 =8). While CD8 associates with MHC Class I (8x1=8). | |
| CD5 | A type of CD which is normally expressed in most T-cells (immature and mature). Recall the CD_Mnemonic. CD5 can also be seen in very small subset of normal B-cells and also aberrantly (abnormally) expressed in certain B-cell Lymphoma/leukemias (e.g. CLL and mantle cell lymphoma). | |
| CD7 | A type of CD which is normally expressed in most T-cells (immature and mature). This is the earliest expressed CD T-cell marker. Recall the CD_Mnemonic | |
| CD8 | A type of CD which is normally expressed in a subset of T-cells (Cytotoxic T-cells). Recall the CD_Mnemonic. This CD is can also be expressed in lower levels on some NK-cells. Also recall the rule of 8 mnemonic: CD4 associates with MCH Class II (4x2 =8). While CD8 associates with MHC Class I (8x1=8). | |
| CD10 | A CD that can be seen expressed in a variety of cell types (hematopoietic and non-hematopoietic). In hematopoietic cells CD10 is commonly expressed in mature neutrophils, early B-cells (hematogones), germinal center B-cells, and certain germinal center derived malignancies such as follicular_lymphoma and Burkitt_lymphoma. | |
| CD11b | Commonly expressed on myelomonocytic cells including granulocytes, monocytes and some AMLs. Recall the CD_Mnemonic. | |
| CD13 | Commonly expressed on myelomonocytic cells including granulocytes, monocytes (brighter expression), and many AMLs. Remember from the CD_Mnemonic, "M" for Middle CDs (CD11 through CD18) and "M" for MyeloMonocytic cells. | |
| CD14 | A CD that is expressed on mature or maturing monocytes. Remember from the CD_Mnemonic, "M" for Middle CDs (CD11 through CD18) and "M" for MyeloMonocytic cells. | |
| CD15 | Commonly expressed on maturing myelomonocytic cells including granulocytes, monocytes and some AMLs. Remember from the CD_Mnemonic, "M" for Middle CDs (CD11 through CD18) and "M" for MyeloMonocytic cells. | |
| CD16 | Commonly expressed on maturing myelomonocytic cells including granulocytes, monocytes and some AMLs. Remember from the CD_Mnemonic, "M" for Middle CDs (CD11 through CD18) and "M" for MyeloMonocytic cells. | |
| CD19 | A type of CD which is normally expressed in most B-cells including normal plasma cells. Recall the CD_Mnemonic, "B" for "B-cells" & "Bigger" CDs (CD19, 20, 21, 22, 23, 24) which are typicaly expressed in "B-cells". Most B-cell Lymphoma/leukemias are CD19 positive. In contrast to normal plasma cells which express CD19, most plasma cell neoplasms (e.g. plasma cell myeloma) do not express CD19. |
|
| CD20 | A type of CD which is normally expressed in most mature B-cells. It is usually negative in early hematogones and normal plasma cells. Recall the CD_Mnemonic, "B" for "B-cells" & "Bigger" CDs (CD19, 20, 21, 22, 23, 24) which are typicaly expressed in "B-cells". Most mature B-cell Lymphoma/leukemias are CD20 positive. CD20 is typically negative in most plasma cell neoplasms (e.g. plasma cell myeloma). CD20 expression on lymphomas is also very important clinically since there is an Anti-CD20 drug (Rituximab) that is typically used as part of the treatment of these lymphomas. | |
| CD21 | Expressed on B-cells and follicular dendritic cells. Co-receptor for Epstein Barr Virus (EBV) which explains why B-cells are typically the cells infected by EBV and not T-cells (T-cells lack CD21). Recall the CD_Mnemonic, "B" for "B-cells" & "Bigger" CDs (CD19, 20, 21, 22, 23, 24) which are typicaly expressed in "B-cells". Note: Unfortunately the function and expression profile of CDs larger than CD24 need to be memorized. |
|
| CD22 | A CD which is normally expressed in most B-cells (both immature and mature) but not typically expressed in plasma cells. Recall the CD_Mnemonic, "B" for "B-cells" & "Bigger" CDs (CD19, 20, 21, 22, 23, 24) which are typicaly expressed in "B-cells". Note: Unfortunately the function and expression profile of CDs larger than CD24 need to be memorized. | |
| CD23 | Expressed on B-cells and follicular dendritic cells. Recall the CD_Mnemonic, "B" for "B-cells" & "Bigger" CDs (CD19, 20, 21, 22, 23, 24) which are typicaly expressed in "B-cells". Note: Unfortunately the function and expression profile of CDs larger than CD24 need to be memorized. | |
| Chediak-Higashi syndrome | Autosomal Recessive disorder leading to a microtubule polymerization defect that reduces phagolysosome formation and phagocytic activity. Hence reduced bacteracidal function. It is characterized by albinism, peripheral neuropathy and increased risk of pyogenic infections. Characteristic neutrophils with chunky large granules.
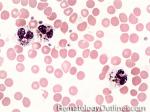 |
|
| Chemotherapy | A medical treatment used in destroying cancer cells. | |
| Chronic | A slow growing or progressing process. The opposite of Acute. (e.g. chronic lymphocytic Leukemia or Chronic inflammation). | |
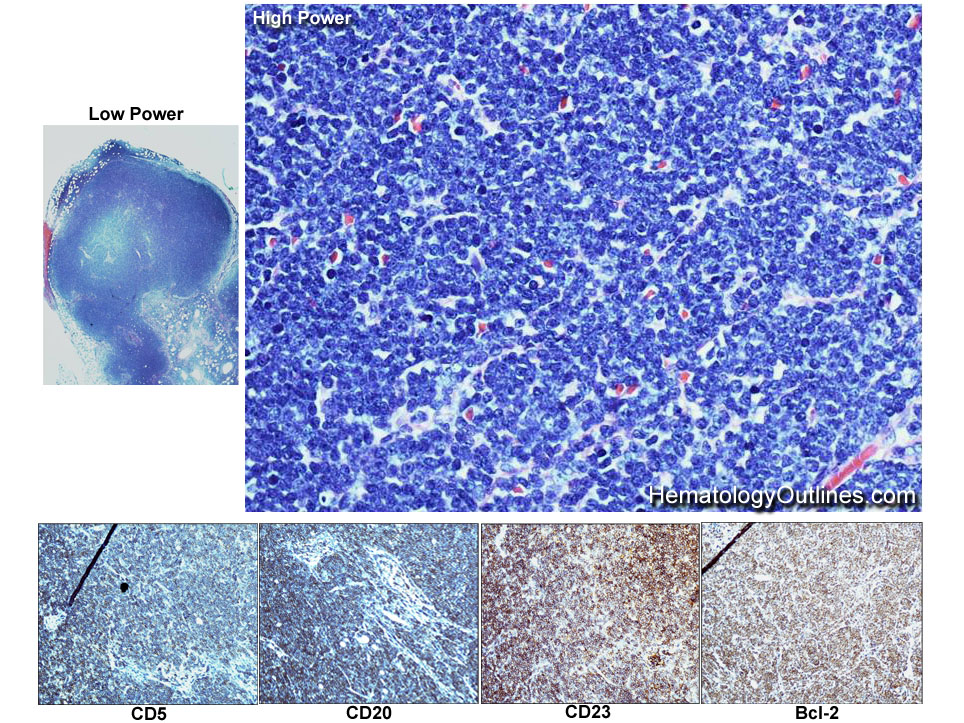
| Chronic lymphocytic leukemia | Also known as CLL. An indolent B-cell Leukemia more common in elderly population. Characteristic immunophenotype (dimCD20+, CD19+, CD5+, CD23+, CD10- monotypic dim kappa or dim lambda B-cells). The disease has two forms: CLL if it's in the blood (hence, "Leukemia") and SLL (Small Lymphocytic Lymphoma) if it's in the tissue such as lymph nodes (hence, "Lymphoma"). Mnemonic: As opposed to Acute Lymphoblastic Leukemia (ALL) which are "immature" cells and more common in "pediatric" (less mature) population, CLL is comprised of "mature" lymphocytes and predominantly seen in elderly (more mature) population.
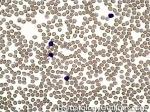  |
|
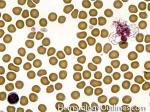
| Chronic myelogenous leukemia | A Myeloproliferative neoplasm of abnormal bone marrow Stem cells that is characterized by the chromosome translocation t(9;22) which leads to a BCR-ABL1 fusion gene (Philadelphia chromosome). Initially presents as an indolent Chronic phase with subsequent transition to accelerated phase and sometimes blast phase. First line therapy usually includes "imatinib" (AKA Gleevec). Similar to most of the other myeloproliferative neoplasms, patients usually present with some elevated count or "cytosis" (such as elevated WBC count which is known as leukocytosis) and splenomegaly. The peripheral blood smear usually shows leukocytosis with increased number of myeloid (granulocytic) precursors in different phases of maturation (Clue: the peripheral blood smear looks like a bone marrow aspirate smear). 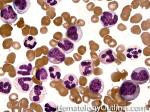 |
|
| Chronic Myelomonocytic Leukemia | AKA CMML is an abnormal bone marrow Stem cell disorder that has characteristic features of both a Myeloproliferative neoplasm ("cytosis" specifically Monocytosis and splenomegaly) and a myelodysplastic syndrome (dysplastic cells). Hence, per WHO 2008 criteria, it belongs to the category of Myeloproliferative/Myelodysplastic disorders. At the time of diagnosis, they usually present with leukocytosis and history of a "persistent Monocytosis" is typically required. As opposed to CML, there is no BCR-ABL1 fusion. | |
| CLL | Also known as Chronic Lymphocytic Leukemia. An indolent B-cell leukemia more common in elderly population. Characteristic immunophenotype (dimCD20+, CD19+, CD5+, CD23+, CD10- monotypic dim kappa or dim lambda B-cells). The disease has two forms: CLL if it's in the blood (hence, "Leukemia") and SLL (Small Lymphocytic Lymphoma) if it's in the tissue such as lymph nodes (hence, "Lymphoma"). Mnemonic: As opposed to Acute Lymphoblastic Leukemia (ALL) which are "immature" cells and more common in "pediatric" (less mature) population, CLL is comprised of "mature" lymphocytes and predominantly seen in elderly (more mature) population.
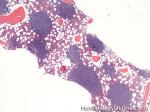 |
|
| Clot | Usually refers to "blood Clot" (AKA Thrombus). The final product of coagulation pathway in hemostasis (platelets and coagulation factors). Normally formed at the site of injury. If exaggerated, it may lead to thrombosis (pathologic). Recall Virchow's Triad which increase risk of thrombosis: 1) Hypercoagulability (e.g. cancer patients), 2) Abnormal blood flow (e.g.due to stasis), & 3) Endothelial injury (e.g.post trauma). | |
| Clotting factors | Proteins within blood plasma involved in the formation and stabalization of the blood clot. Common examples include factors I, II, V, VII, VIII, IX, X, and XI. | |
| Cluster of Differentiation | AKA "CD". These are glycoproteins expressed by normal and abnormal cells and can be used as part of the diagnostic workup in many hematologic malignancies by either flow cytometry or immunohistochemistry (e.g. co-expression of CD5 and CD23 on CLL cells). | |
| CML | Chronic Myelogenous Leukemia. A Myeloproliferative neoplasm of abnormal bone marrow Stem cells that is characterized by t(9;22) which leads to a BCR-ABL1 fusion gene (Philadelphia chromosome). Initially presents as an indolent chronic phase with subsequent transition to accelerated phase and sometimes blast phase. First line therapy usually includes "imatinib" (AKA Gleevec). Similar to most of the other myeloproliferative neoplasms, patients usually present with some "cytosis" (such as elevated WBC which is known as leukocytosis) and splenomegaly. The peripheral blood smear usually shows leukocytosis with increased number of myeloid (granulocytic) precursors in different phases of maturation (Clue: the peripheral blood smear looks like a bone marrow aspirate smear). |
|
| CMML | AKA Chronic MyeloMonocytic Leukemia is an abnormal bone marrow Stem cell disorder that has characteristic features of both a Myeloproliferative neoplasm ("cytosis" specifically Monocytosis and splenomegaly) and a myelodysplastic syndrome (dysplastic cells). Hence, per WHO 2008 criteria, it belongs to the category of Myeloproliferative/Myelodysplastic disorders. At the time of diagnosis, they usually present with leukocytosis and history of a "persistent Monocytosis" is typically required. As opposed to CML, there is no BCR-ABL1 fusion.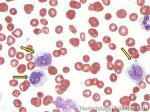 |
|
| Coagulopathy | This usually refers to a bleeding or clotting disorder which is typically secondary to an impairment of the blood's ability to form a thrombus (clot). Mostly the defect or deficiency of the coagulation factors leads to an increased risk of bleed (e.g. hemophilia A due to factor VIII deficiency or vW disease due to a defect or deficiency of vW factor) but sometimes the deficiency may lead to an increased risk of thrombosis (clottting) such as in Factor XII deficiency. | |
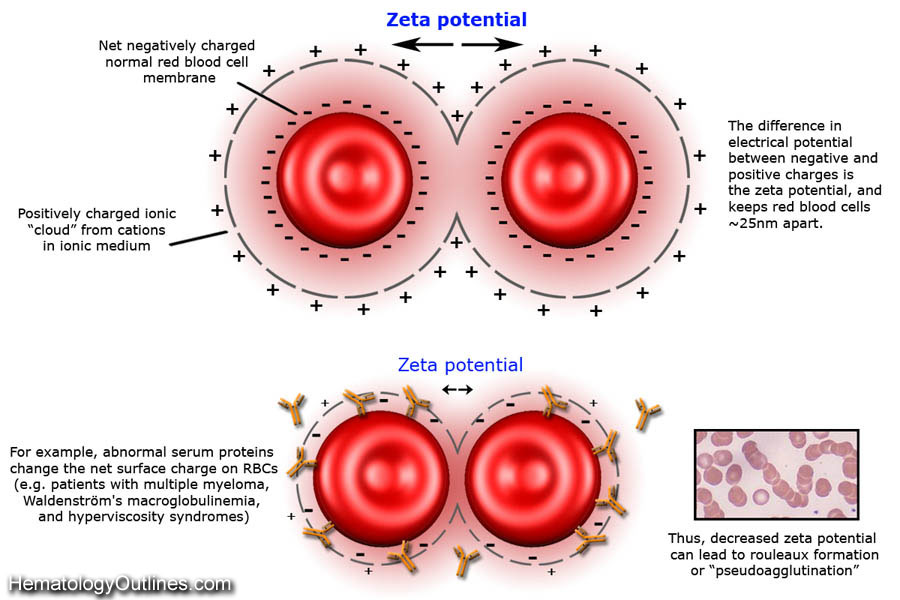
| Cold agglutinins | Usually associated with a cold-reacting IgM antibody that is usually not clinically significant at body temperature. On peripheral smear you may see RBC agglutination (Clumping of RBCs). However, cold agglutinins may cause cold agglutinin disease. | |
| Complete Blood Count | Also known as CBC which includes a count of the WBC, hemoglobin, hematocrit and platelets. When a differential count is added to the CBC, the different types of WBCs are quantitated (e.g. percent neutrophils, lymphocytes, monocytes, eosinophils and basophils). | |
| Coombs test | AKA Antiglobulin test. Two types: Direct Antiglobulin Test (DAT) and Indirect Antiglobulin Test (IAT). Direct detects Abs or complement bound to patient's RBCs while Indirect detects Abs against RBCs in the patient's Serum (plasma). An example of DAT positive disease is Autoimmune Hemolytic anemia. An example of IAT is detecting Alloantibodies in the patient's plasma prior to transfusing them with a specific type of RBC (e.g. Kell negative RBC). | |
| Coumadin | Generic name: Warfarin is an anticoagulant that acts by inhibiting the Vitamin K-dependent synthesis of factors II, VII, IX, and X, as well as proteins C & S. Used in the prevention of thrombosis and clinical settings such as atrial fibrillation, the presence of artificial heart valves, deep venous thrombosis, and pulmonary embolism. Degree of anticoagulation is monitored by INR. Coumadin is contraindicated in pregnancy. Adverse effects include hemorrhage, warfarin necrosis, and osteoporosis. Effects of Coumadin can be reversed by giving vitamin K. Fresh-frozen plasma (FFP) can be given when rapid reversal is needed. | |
DBack to top ^ | ||
| D-Dimer | Products of a blood clot degeneration or breakdown. Can be the result of plasmin degrading fibrin into D-Dimer (Two Cross-linked D fragments of fibrinogen protein). Hence it's a type of FDP. Usually increased after thrombotic events and disseminated intravascular coagulation (DIC). Note: D-Dimer levels can be used to exclude thrombosis if the probability of thrombosis is low and the D-Dimer is negative (good negative predictive value). |
|
| Delta granules | AKA Dense bodies are secretory organelles in platelets containing ADP, ATP, Calcium & Serotonin. Compared to alpha granules, these are all smaller molecules. Mnemonic is "Delta or Dense" granules are Diminutive (tiny molecules) as opposed to alpha granules. | |
| Deoxyhemoglobin | Hemoglobin that is not bound to oxygen (Oxygenated hemoglobin unloads oxygen in tissue and becomes deoxyhemoglobin). | |
| DIC | Disseminated Intravascular Coagulation is a microangiopathic hemolytic anemia that is characterized by thrombocytopenia and consumption of coagulation factors (i.e. there is usually an increase in both PT and PTT lab tests). DIC is not a disease by itself but rather secondary to an underlying disease process (e.g. secondary to infection, malignancy, trauma, etc.). On peripheral smear you may see increased numbers of schistocytes which are caused by fibrin-induced intravascular hemolysis. | |
| Disseminated Intravascular Coagulation | AKA DIC is a microangiopathic hemolytic anemia that is characterized by thrombocytopenia and consumption of coagulation factors (i.e. there is usually an increase in both PT and PTT lab tests). DIC is not a disease by itself but rather secondary to an underlying disease process (e.g. secondary to infection, malignancy, trauma, etc.). On peripheral smear you may see increased numbers of schistocytes which may have been caused by a fibrin-induced intravascular hemolysis. | |
| Dohle bodies | Blue-Gray cytoplasmic inclusions (remnants of Rough ER) that may be seen in neutrophils. Increased numbers may be indicative of an underlying inflammatory condition and/or infection. If Dohle bodies are accompanied by giant platelets, it may be due to May Hegglin anomaly.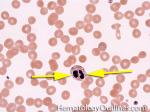 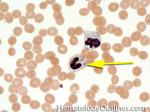 |
|
| dRVVT | AKA Dilute russell viper venom time is an in vitro qualitative test for lupus anticoagulant (LA) and usually follows an elevated PTT when LA is suspected. The test is derived from the venom of Russell viper, a power thrombotic agent in vitro. A mixing study is performed combining Russell viper venom, patient’s plasma, and phospholipids, which is required for coagulation. The presence of lupus anticoagulant would prevent clotting. A prolonged clotting time would be followed up with a confirmatory test where excess phospholipids are added to the mixing study, which should overcome LA and induce clotting. A ratio between clotting time without excess phospholipids and with excess phospholids is then calculated. An elevated ratio is considered positive and consistent with LA. | |
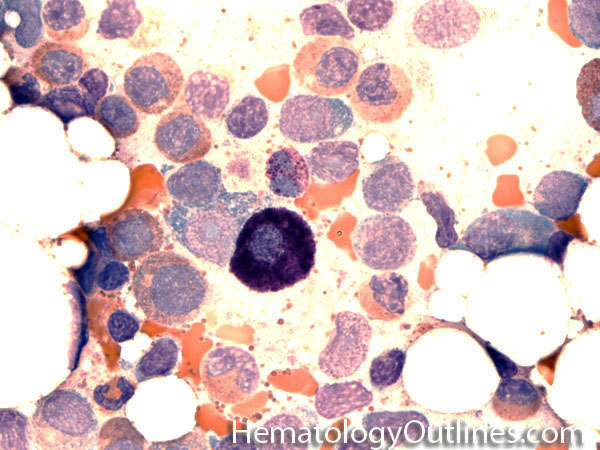
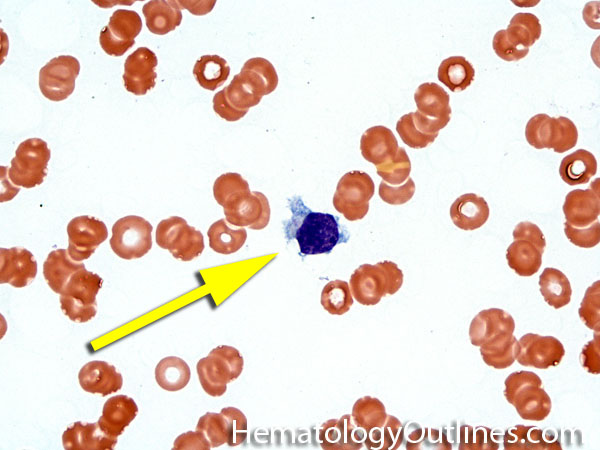
| Dutcher bodies | Intranuclear pseudoinclusions noted in plasma cells. Usually associated with abnormal plasma cells such as plasmacytoma or plasma cell myeloma. | |
| Dys | Dys means abnormal. Examples include Dyserythropoiesis (abnormal erythroid maturation), dysplasia (abnormal growth or abnormal formation), etc. | |
EBack to top ^ | ||
| EBV | Epstein Barr Virus. A virus associated with infectious mononucleosis (a disease more common in younger patients that presents with lymphadenopathy and in severe cases with organomagly such as hepatomegaly and/or splenomegaly). EBV may also be associated with certain lymphoproliferative disorders such as Post Transplant lymohoproliferative disorders (PTLD), angioimmunoblastic T-cell Lymphoma and Hodgkin lymphoma. | |
| Eculizumab | Brand name: Soliris is a recombinant monoclonal antibody directed against complement protein C5. It inhibits the cleavage of C5 by C5 convertase which prevents the generation of the membrane attack complex, C5b-C9. Eculizumab is used to treat paroxysmal nocturnal hemoglobinuria (PNH) which is a disease characterized by complement-mediate intravascular hemolysis. This drug has also been used to treat certain hemolytic uremic syndrome (HUS) cases. | |
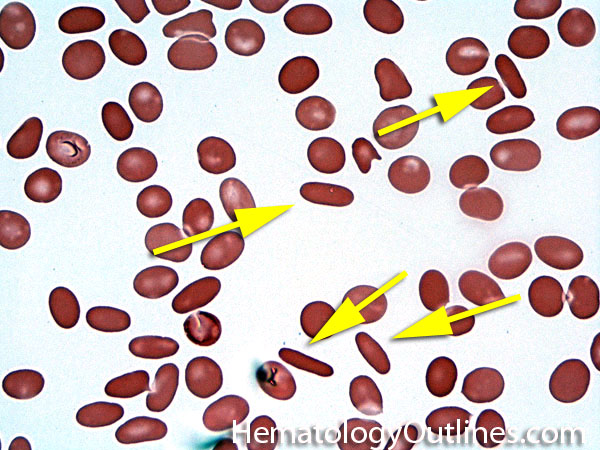
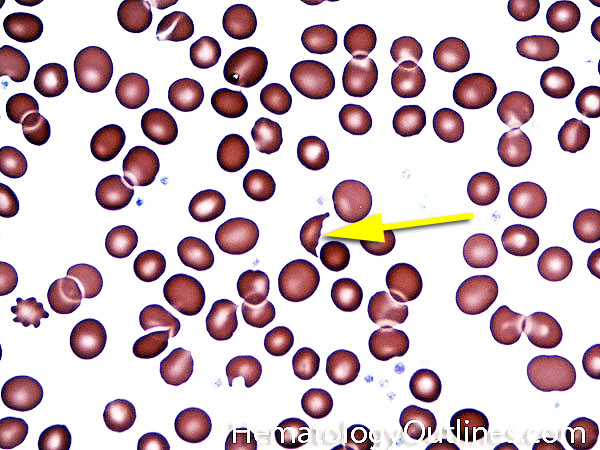
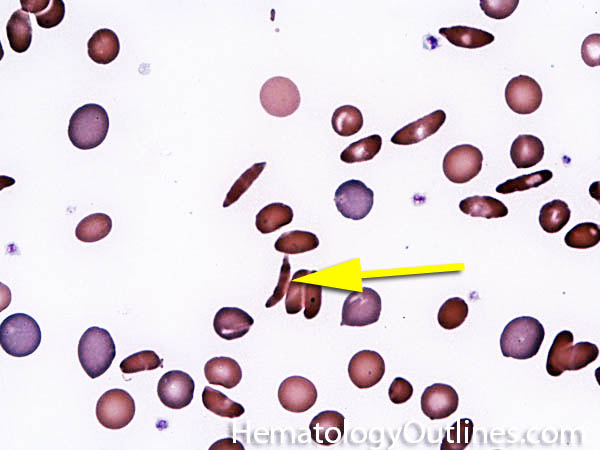
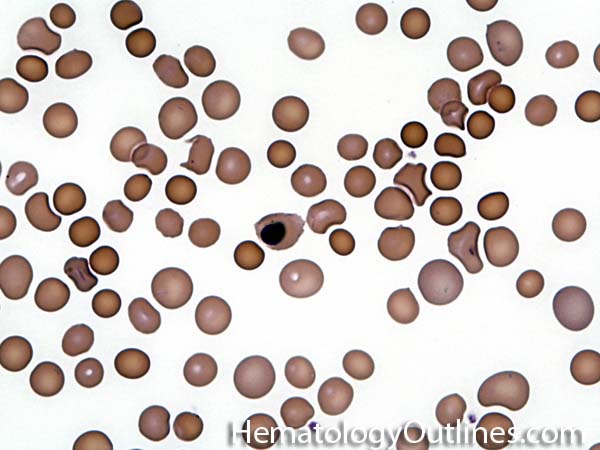
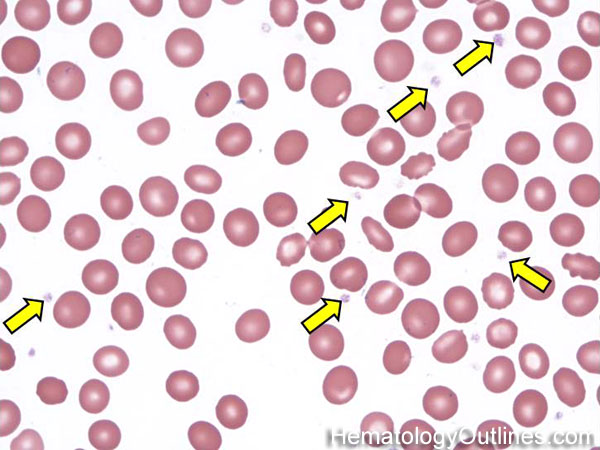
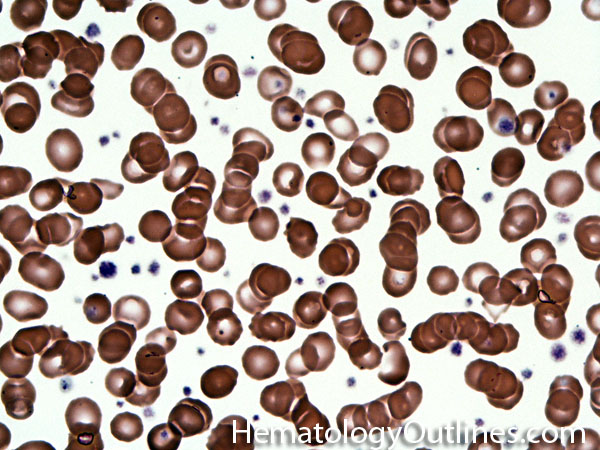
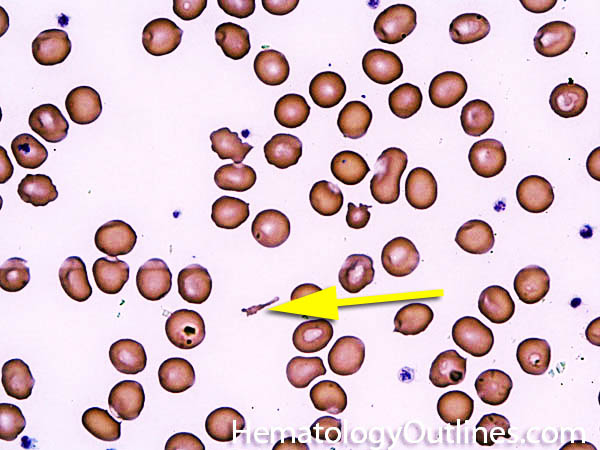
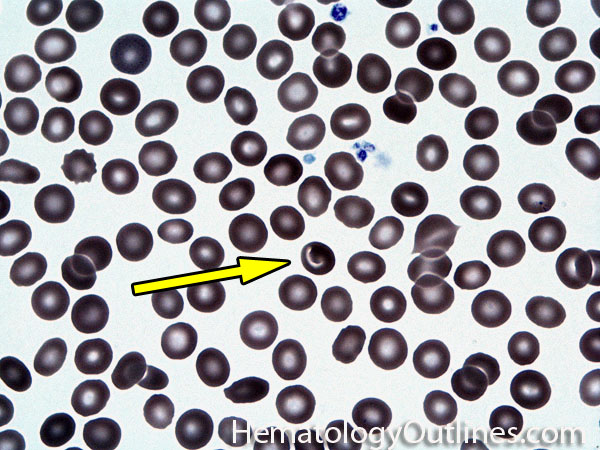
| Elliptocytes | Oval shaped RBCs (AKA ovalocytes). Can be seen in variety of conditions including iron deficiency anemia and hereditary elliptocytosis.
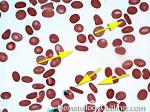  |
|
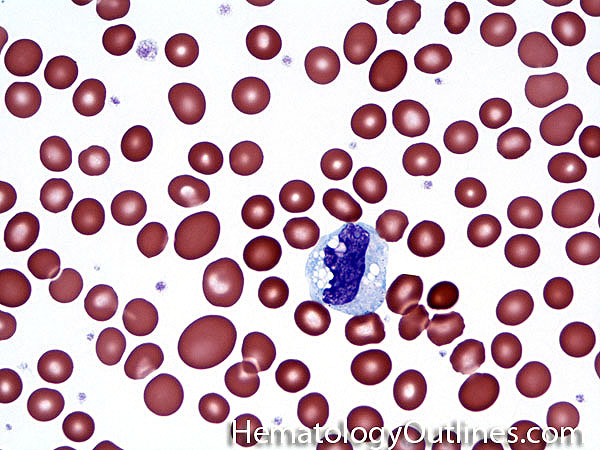
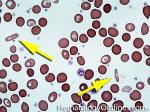
| Eosinophils | A type of WBC seen in peripheral blood and tissue. It belongs to the myeloid lineage and more specifically a subtype of granulocyte with bright eosinophilic (red) granules containing Major Basic Protein, histamine, and plasminogen. Increased numbers of eosinophils in blood can be due to many things (remember the major classes for most differential diagnosis include an underlying Infection, Malignancy, autoimmune, trauma, congenital, drug-induced or idiopathic). Increased eosinophils may be due to an infection (e.g. tissue invasive parasites), Malignancy (e.g. Hodgkin Lymphoma, Carcinoma or Chronic Eosinophilic Leukemia), Autoimmune, Drug-induced (e.g. certain antibiotics, etc.), or idiopathic.
  |
|
| Eosinophilia | Refers to increased number of eosinophils in the blood. DDx: Secondary to a tissue invasive parasitic infection, Autoimmune, Malignancy, drugs, or idiopathic).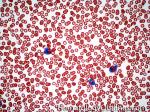 |
|
| EPO | a.k.a erythropoietin is a glycoprotein hormone that stimulates erythropoiesis. It is mainly produced by the liver during the fetal period and by the kidney in adults. Manufactured EPO is typically used to treat anemia due to Chronic renal disease, myelodysplasia, and anemia associated with cancer treatment (post chemotherapy and radiation). | |
| Erythrocyte | Another name for Red blood Cell (RBC). Belongs to the myeloid cell lineage. RBCs main role is to carry and deliver oxygen through hemoglobin.
|
|
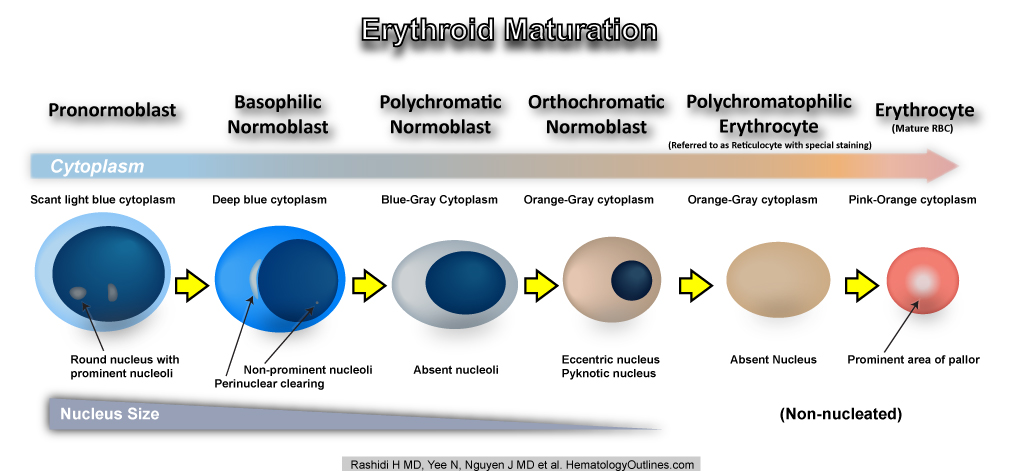
| Erythroid maturation | Erythroids (RBCs) start as nucleated cells in the marrow and ultimately lose their nucleus prior to leaving the marrow and entering the blood. (see image).
 |
|
| Erythrophagocytosis | Refers to RBCs ingested (phagocytosed) by macrophages and sometimes other WBCs. | |
| Erythropoietin | a.k.a EPO is a glycoprotein hormone that stimulates erythropoiesis. It is mainly produced by the liver during the fetal period and by the kidney in adults. Manufactured EPO is typically used to treat anemia due to Chronic renal disease, myelodysplasia, and anemia associated with cancer treatment (post chemotherapy and radiation). | |
FBack to top ^ | ||
| FAB classification | FAB stands for French, American, British which was the old classification scheme used in classifying certain hematologic malignancies such as Acute Myeloid Leukemias (AMLs M0 through M7), acute lymphoblastic Leukemia (ALL) and Myelodysplastic Syndromes (MDS). | |
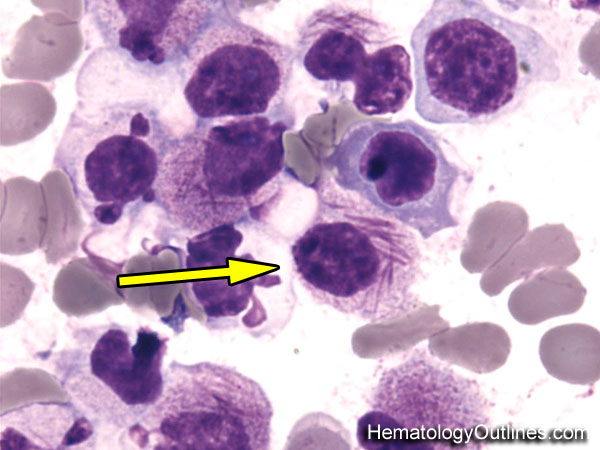
| Faggot cell | Collection of Auer rods sometimes seen within blasts in Acute Promyelocytic Leukemia (APL). |
|
| FDP | Fibrin degradation products (FDP) are the result of blood clot degeneration or breakdown. Can be the result of plasmin degrading fibrin into FDPs (e.g. D-Dimer which is a type of FDP: Two Cross-linked D fragments of fibrinogen protein). FDPs are increased after thrombotic events and disseminated intravascular coagulation (DIC). Note: D-Dimer (a type of FDP) levels can be used to exclude thromobosis if the probability of thrombosis is low and the D-Dimer is negative. | |
| Ferritin | A protein that is present in many cell types and serves to store Iron. It is also an Acute Phase Protein meaning that it's levels may rise in a background of inflammation or stress. | |
| Fibrin degradation product | Also known as FDP are products of a blood clot degeneration. Can be the result of plasmin degrading fibrin into FDPs (e.g. D-Dimer: Two Cross-linked D fragments of fibrinogen protein). FDPs are increased after thrombotic events and disseminated intravascular coagulation (DIC). Note: D-Dimer (a type of FDP) levels can be used to exclude thromobosis if the probability of thrombosis is low and the D-Dimer is negative. | |
| Fibrinogen | a.k.a Factor I. Glycoprotein synthesized in the liver that is involved in coagulation cascade. Fibrinogen is cleaved by thrombin to form fibrin. Fibrin is then cross-linked by Factor XIII to form a clot. Note: Fibrinogen is also an Acute-phase reactant which is elevated during inflammation. | |
| Flow cytometry | A laboratory test that characterizes cells based on their immunophenotype by using antibodies that tag cell surface or nuclear epitopes. It is analogous to the concept of tissue immunohistochemistry (IHC). Advantages of flow cytometry over IHC are: quicker turn-around time, being able to evaluate multiple antibodies on a cell population simultaneously, etc. It is commonly used in diagnostic hematology to characterize hematologic malignancies (Acute Leukemias, Lymphomas, etc.). | |
| Fludarabine | Brand name: Fludara is a chemotherapy drug used to treat Chronic lymphocytic Leukemia (CLL), salvage therapy of non-Hodgkin Lymphoma, and Acute myeloid leukemias (AML). Side effects include anemia, thrombocytopenia, neutropenia, and profound lymphopenia. | |
| Folate Deficiency | Folate a.k.a folic acid is a water-soluble vitamin with an important role in DNA synthesis and repair. It is typically given during pregnancy to prevent neural tube defects in the embryo. Deficiency in folate or folic acid can also result in macrocytic anemia. Methotrexate, a drug that interferes with folate metabolism, inhibits the production of tetrahydrofolate (THF), an active form of folic acid, and can cause inflammation in the GI tract and marrow suppression. Therefore, folate supplement is usually given to reverse the toxic side effects of methotrexate. Folic acid, together with Vitamin B12, is important in the conversion of homocysteine to methionine, therefore folate and/or vitamin B12 deficiency can lead to hyperhomocyteinemia (see vitamin B12). However, as opposed to B12 deficiency, MMA is not elevated in Folate deficiency since it is not involved in the conversion of methylmalonyl-CoA to succinyl-CoA in the Kreb cycle. Take home points: Folate deficiency may show hyperhomocyteinemia but no elevation in MMA (Methyl Malonic Acid). While B12 deficiency may show both hyperhomocyteinemia and an elevated MMA. | |
| Follicular lymphoma | A common mature B-cell Lymphoma that usually shows a nodular growth pattern and more commonly behaves in a low grade fashion. Immunophenotypically the neoplastic B-cells in this lymphoma are CD19+ and CD20+ ("Big" CDs meaning "B" cell origin), CD10+ and BCL2+. The common translocation associated with this lymphoma is t(14;18) which includes IgH and BCL2 genes, see Lymphoma_mnemonic.
  |
|
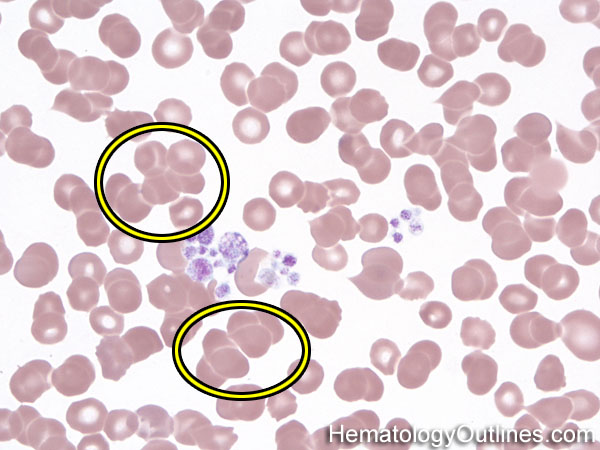
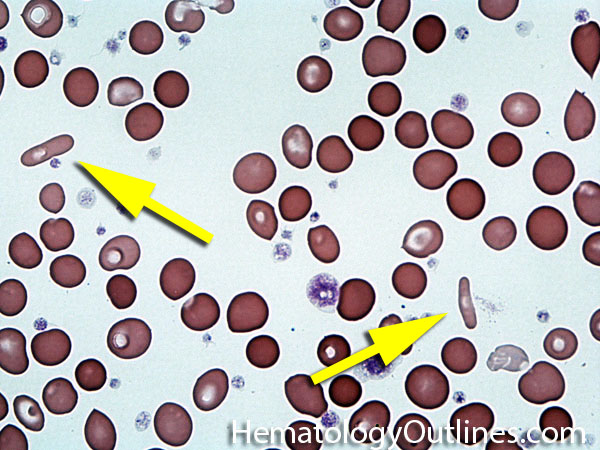
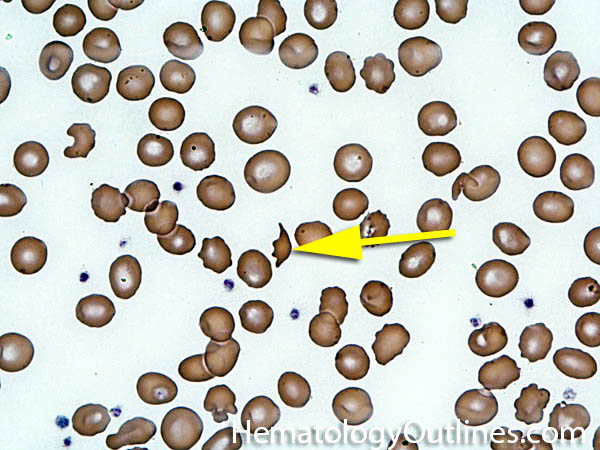
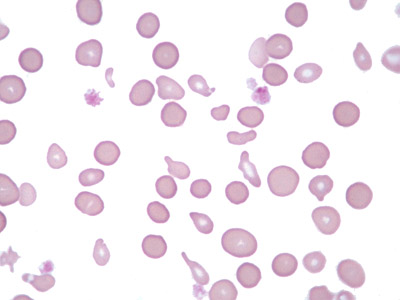
| Fragmented red blood cells | AKA schistocytes and helmet cells are irregularly shaped assymetrical fragments of RBCs that may have several morphologic forms. It is usually the result of mechanical disruption of the RBCs. They can be generated by getting stuck to fibrin strands within the vasculature secondary to an underlying Microangiopathic hemolytic anemia such as HUS, TTP and DIC or broken up by a mechanical heart valve). 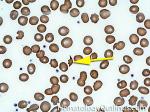 |
|
GBack to top ^ | ||
| G6PD deficiency | An enzyme deficiency that makes RBCs more susceptible to oxidative damage. This oxidative damage is in the hemoglobin molecules which precipitates out as Heinz bodies which are then deposited on RBC membrane and can be phagocytized by splenic macrophages leading to bite cells (RBCs that look like someone has taken a bite out of them). Conditions that may increase the chance of oxidative damage in these patients include infections, certain Sulfa drugs, or fava beans.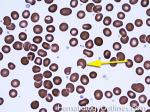 |
|
| GCSF | Generic name: Filgrastim, Brand name: Neupogen is a Granulocyte colony-stimulating factor, a growth factor hormone that stimulates the bone marrow to produce granulocytes and stem cells. It is used to accelerate recovery from neutropenia after chemotherapy and as a supportive medication used to prevent infection and neutropenic fevers. GCSF is also used to increase the number of hematopoietic stem cells in the blood of donors before collection by leukopheresis for use in hematopoietic stem cell transplantation. | |
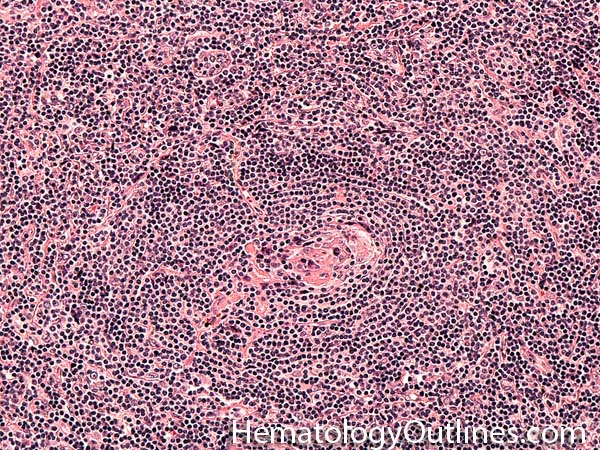
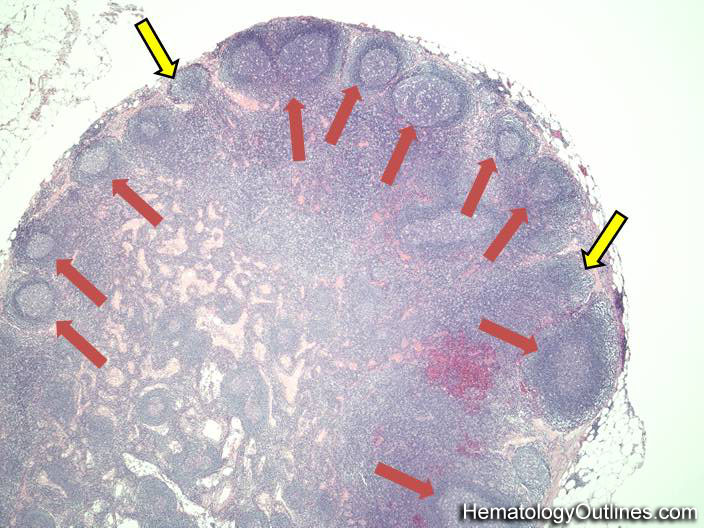
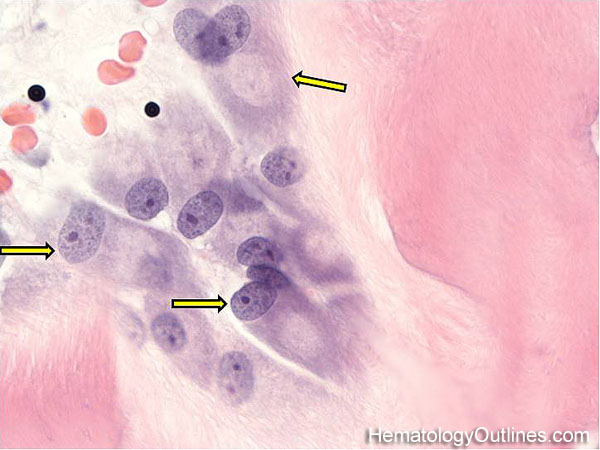
| Germinal center | These are the central portion of the secondary follicles in lymphoid tissue (e.g. lymph node, MALT, or Spleen). As the primary follicle (no germinal center) gets antigen stimulated, the primary follicle then turns into a secondary follicle (germinal center is in the middle of these secondary follicles). Upon antigen stimulation, B-cells within these germinal centers continue their maturation cycle by undergoing somatic hypermuatation and heavy chain class switching which could ultimately lead to the development of a plasma cell or memory B-cell.  |
|
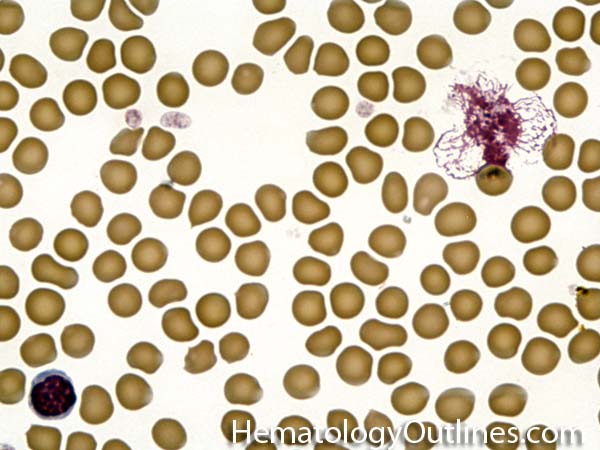
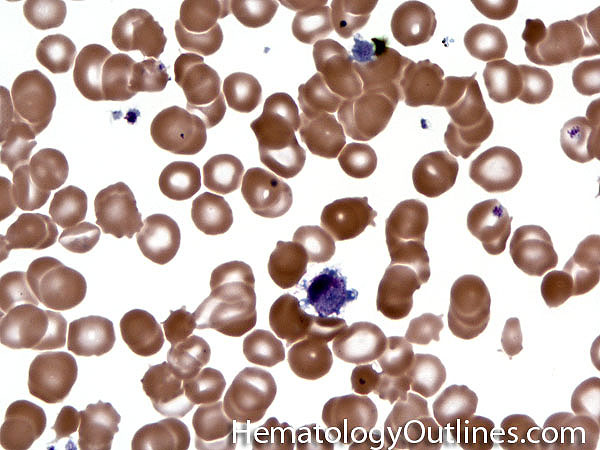
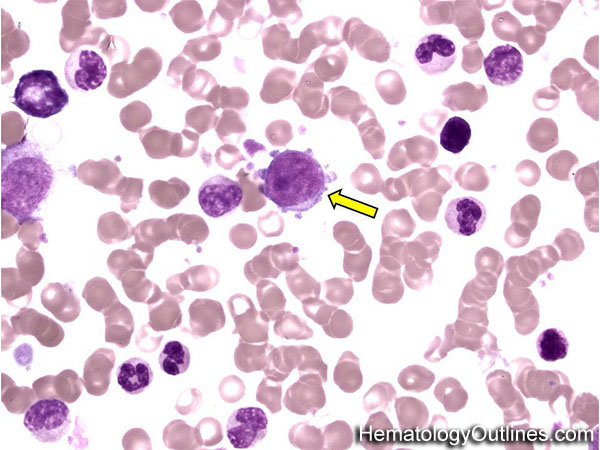
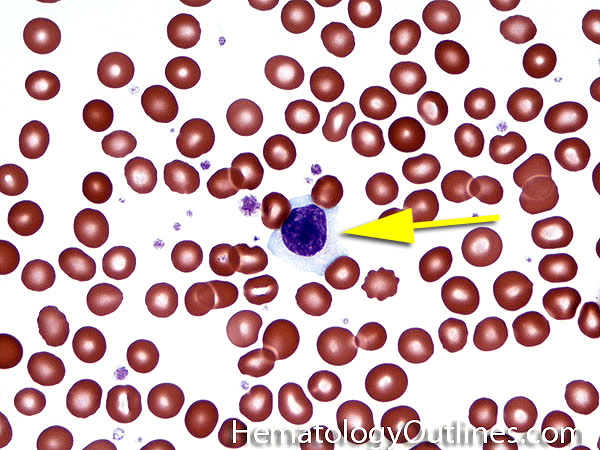
| Giant platelets | Very large platelets. Usually equal to or larger than the size of a normal RBC. Increased numbers in peripheral blood smears may be indicative of an underlying marrow or myeloid disorder.
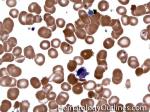  |
|
| Globin chain | This usually refers to one of the peptide chains that comprise the hemoglobin molecule. There are multiple types of globin chains with various combinations that comprise different hemoglobin molecules. These include the alpha, beta, gamma and delta chains. Combination of these globin chains gives rise to multiple types of normal hemoglobin molecules. In order of most to least prevalent Hemoglobins, Hemoglobin A (HbA) is comprised of 2 Alphas + 2 Beta chains, Hemoglobin A2 (HbA2) is comprised of 2 Alphas + 2 Delta chains, and Hemoglobin F (HbF) is comprised of 2 Alphas + 2 Gamma chains. | |
| Glucocorticoids | A class of steroid hormones produced by the adrenal cortex and involved in immune regulation and glucose metabolism by acting through glucocorticoid receptors. An example is cortisol, produced by the zona fasiculata in the adrenal cortex, which regulates blood sugar through gluconeogenesis, suppresses the immune system, and aids in fat, protein and carbohydrate metabolism. Synthetic form is manufactured and used to replace physiologic deficits, such as in adrenal insufficiencies, produce therapeutic immunosuppression, and as anti-inflammatory modulators. | |
| Granulation tissue | A reparative process in tissues that is usually post-injury. Eventually, the goal of the injured tissue is to form a granulation tissue and ultimately recruit fibroblasts and lead to fibrosis. Not to be confused with granuloma (granulomatous inflammation) which by definition is a collection of epithelioid histiocytes. | |
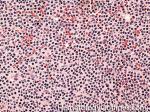
| Granulocytes | Belongs to the myeloid lineage. There are three different types: neutrophils (with neutral staining granules), eosinophils (with Eosinophilic or red staining granules), and basophils (with basophilic or blue/purple staining granules). 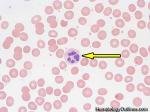 |
|
| Granuloma | AKA granulomatous inflammation is a collection of epithelioid histiocytes (large histiocytes with increased cytoplasm). May be due to an underlying infection (e.g. Mycobacterial or fungi), Malignancy (e.g. Hodgkin Lymphoma), or other processes (e.g. Sarcoidosis). Not to be confused with granulation tissue.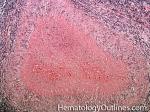 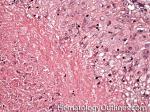 |
|
| Granulomatous Inflammation | AKA granuloma is a collection of epithelioid histiocytes (large histiocytes with increased cytoplasm). May be due to an underlying infection (e.g. Mycobacterial or fungi), Malignancy (e.g. Hodgkin Lymphoma), or other processes (e.g. Sarcoidosis). Not to be confused with granulation tissue. |
|
| Gray platelet syndrome | AKA Alpha Granule Deficiency is a rare inherited bleeding disease that is due to loss of alpha granules in platelets. Recall, alpha granules contain the larger molecules such as PDGF, PF-4, Fibronectin and vWF (as opposed to delta or dense granules that contain the smaller molecules such as serotonin, ADP, ATP and Calcium). | |
HBack to top ^ | ||
| H bodies | AKA hemoglobin H is an abnormal hemoglobin (Beta chain tetramer) that is the result of excess B-chain production due to underproduction of alpha chains in severe alpha thalassemia. | |
| Hairy cell leukemia | An indolent B-cell Leukemia with leukemic cells (abnormal cells circulating in the blood) that have cytoplasmic projections resembling "Hair-like" projections. Patients usually present with some cytopenia and splenomegaly with the neoplastic cells involving the red pulp of the spleen. The typical immunophenotype of hairy cell is CD19+, CD20+, CD25+, CD103+ and CD11c+.
 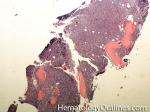 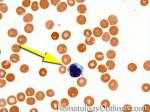 |
|
| Haptoglobin | A serum protein that can bind free hemoglobin. Hence, after intravascular hemolysis, the free haptoglobin binds the released hemoglobin molecules so the levels of the measured free haptoglobin will fall. Note: A low haptoglobin lab value could be secondary to an intravascular hemolysis or sometimes due to severe extravascular hemolysis. | |
| Hb | Abbreviation of hemoglobin. A major protein in Red blood cells (RBCs) that is responsible for carrying oxygen. It is composed of four peptide chains. The three major normal hemoglobins are Hb-A ( 2 alpha chains and 2 Beta chains), Hb-A2 (2 alpha chains and 2 delta chains), and Hb-F (2 alpha chains and 2 gamma chains). Hemoglobin can be measured along with Hct, WBC and platelets as part of a routine CBC (complete blood count). Decreased hemoglobin and Hct levels are indicative of anemia. | |
| Hct | Abbreviation of hematocrit. A percentage of blood that is taken up by RBCs. Low levels is usually indicative of anemia. The normal ratio of hemoglobin to hematocrit is about 1:3 (e.g. Hemoglobin of 10 g/dL usually coorelates with a Hematocrit of 30%). | |
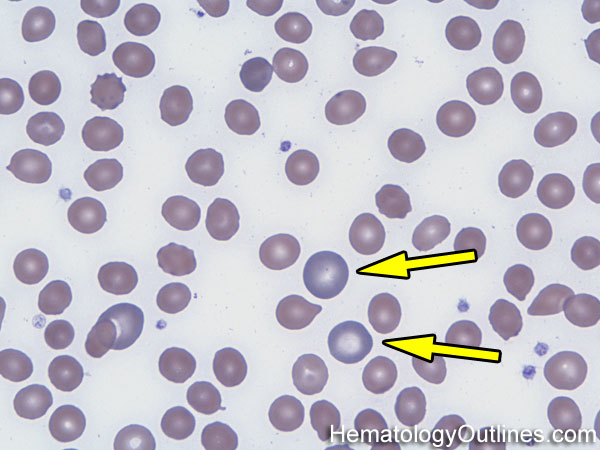
| Heinz bodies | Abnormal preciitated hemoglobins usually due to oxidative damage. These may be seen in conditions that predispose patient's hemoglobin to oxidative damage (e.g. G6PD deficiency). Remember "HEinz" start with HE which stands for HEmoglobin (as opposed to Howell Jolly Bodies which are DNA remnants).
|
|
| Helmet cell | AKA schistocyte is an irregularly shaped asymmetrical fragment of RBC that may have several morphologic forms. It is usually the result of mechanical disruption of the RBCs. They can be generated by getting stuck to fibrin strands within the vasculature secondary to an underlying Microangiopathic hemolytic anemia such as HUS, TTP and DIC or broken up by a mechanical heart valve).
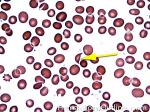 |
|
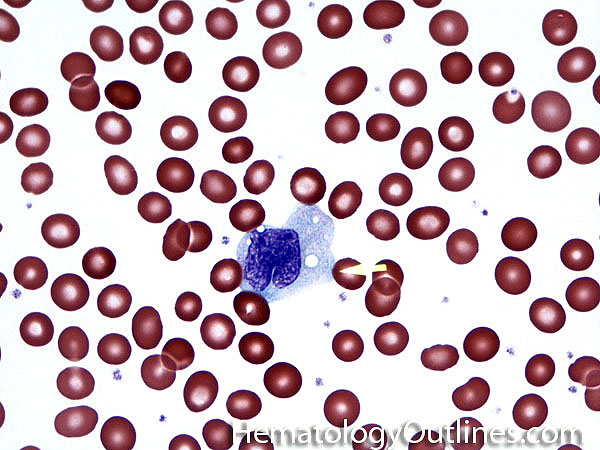
| Helper T cell | These are a type of T-cell lymphocyte that express CD4 and do not have cytotoxic or phagocytic capabilities. Increased numbers of CD4+ T-cells could be due to a maligancy such as T-cell Lymphoma (neoplastic T-cells) or Hodgkin lymphoma (non-neoplastic background T-cells), certain infections such as Mycobacterium tuberculosis or fungi with granulomatous inflammation (granuloma). | |
| Hematocrit | AKA Hct. A percentage of blood that is taken up by RBCs. Low levels is usually indicative of anemia. The normal ratio of hemoglobin to hematocrit is about 1:3 (e.g. Hemoglobin of 10 g/dL usually coorelates with a Hematocrit of 30%). | |
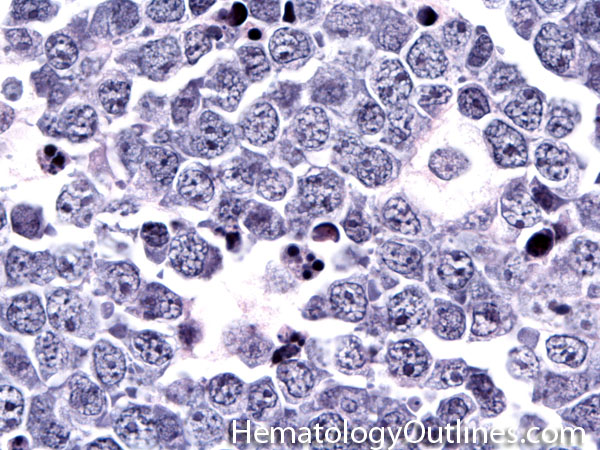
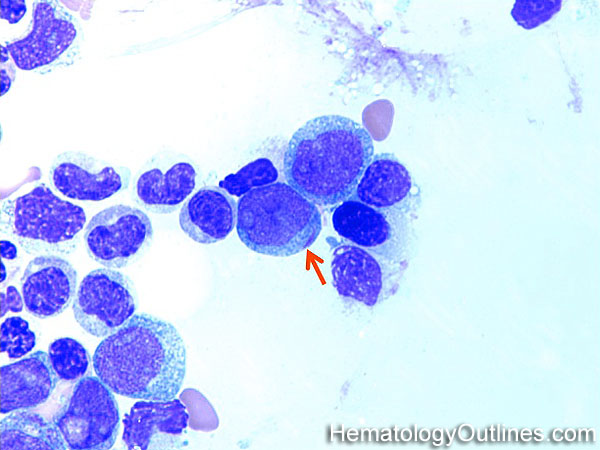
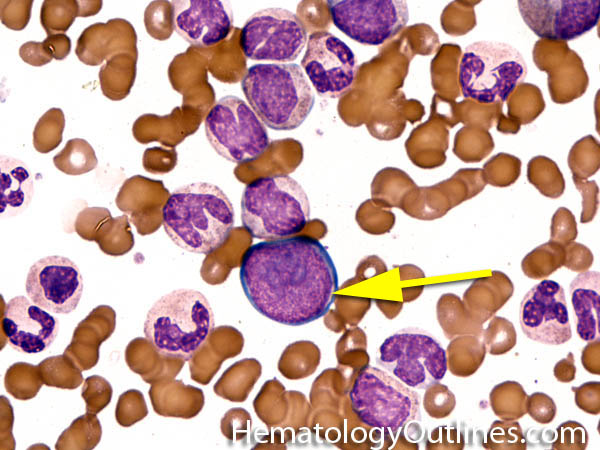
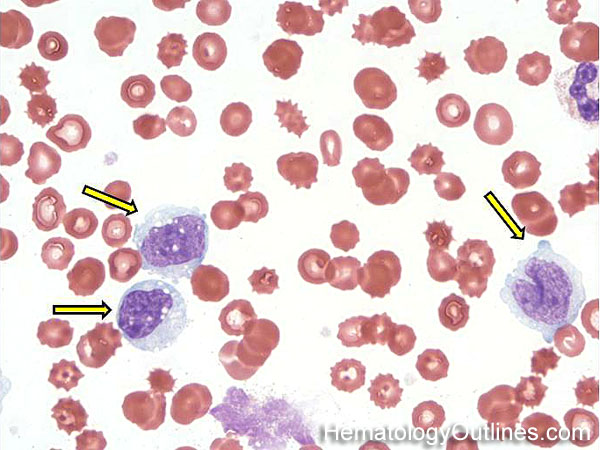
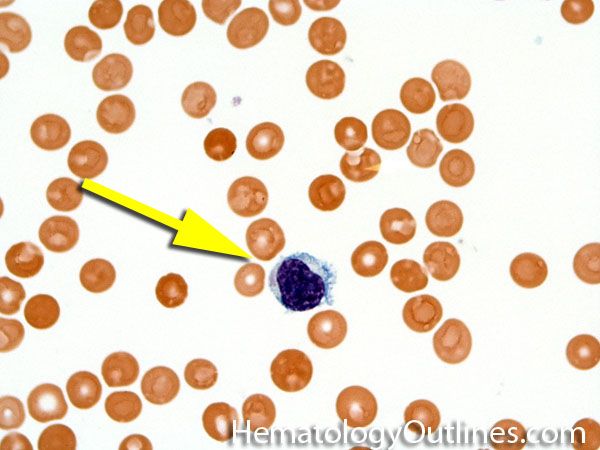
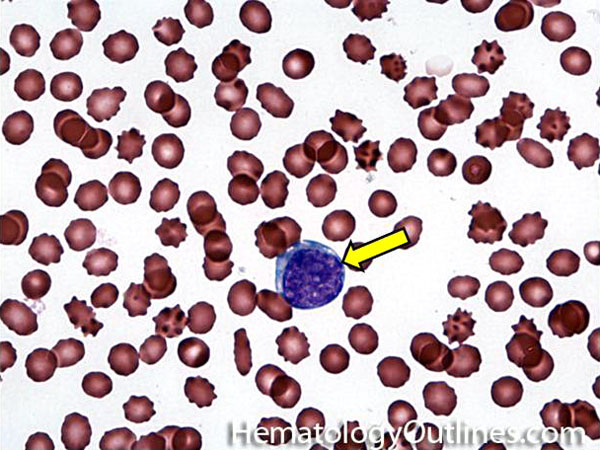
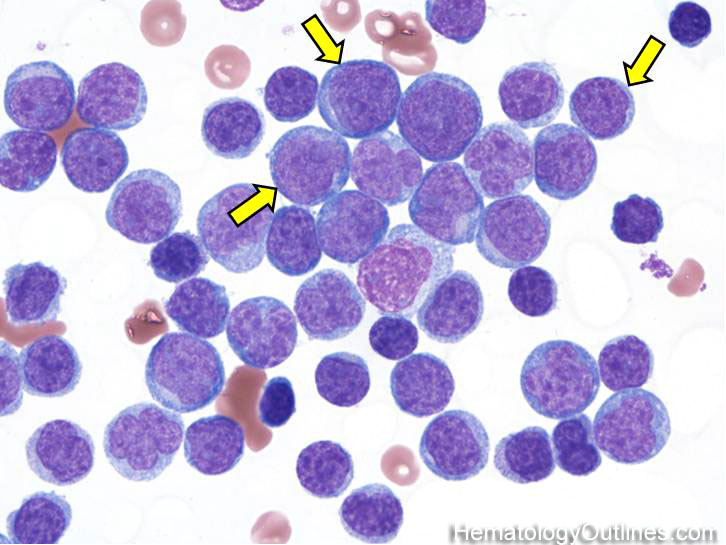
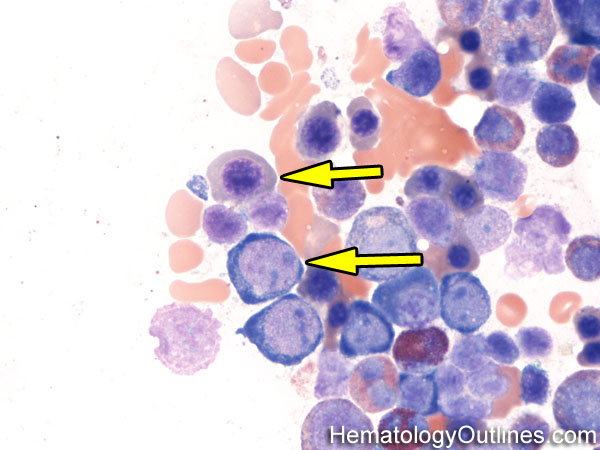
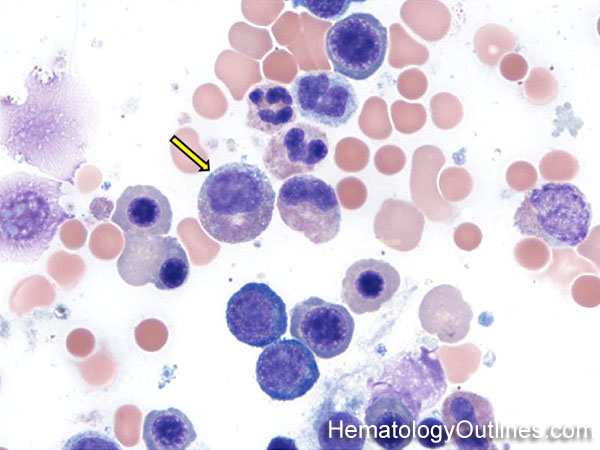
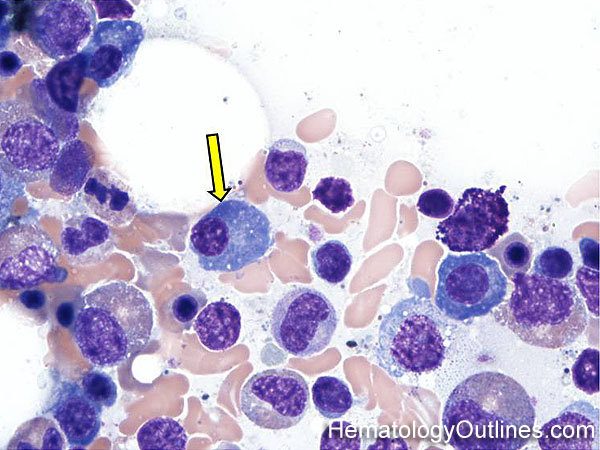
| Hematogones | Early B-cells in the developmental stage following precursor B-cells and before small naïve B-cell found in the bone marrow. Hematogones are more abundant in normal infant marrow and less common in normal adult marrow. A normal hematogone population shows a range of morphology and immunophenotype reflecting a spectrum of maturation. The typical morphology of hematogones is CD19+ and CD10+ with variable expression of CD34, TdT and CD20 depending on their maturation stage. They typically do not express Kappa or Lambda light chains.
(Early Hematogones)
(Late Hematogones)
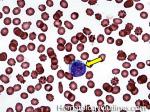 |
|
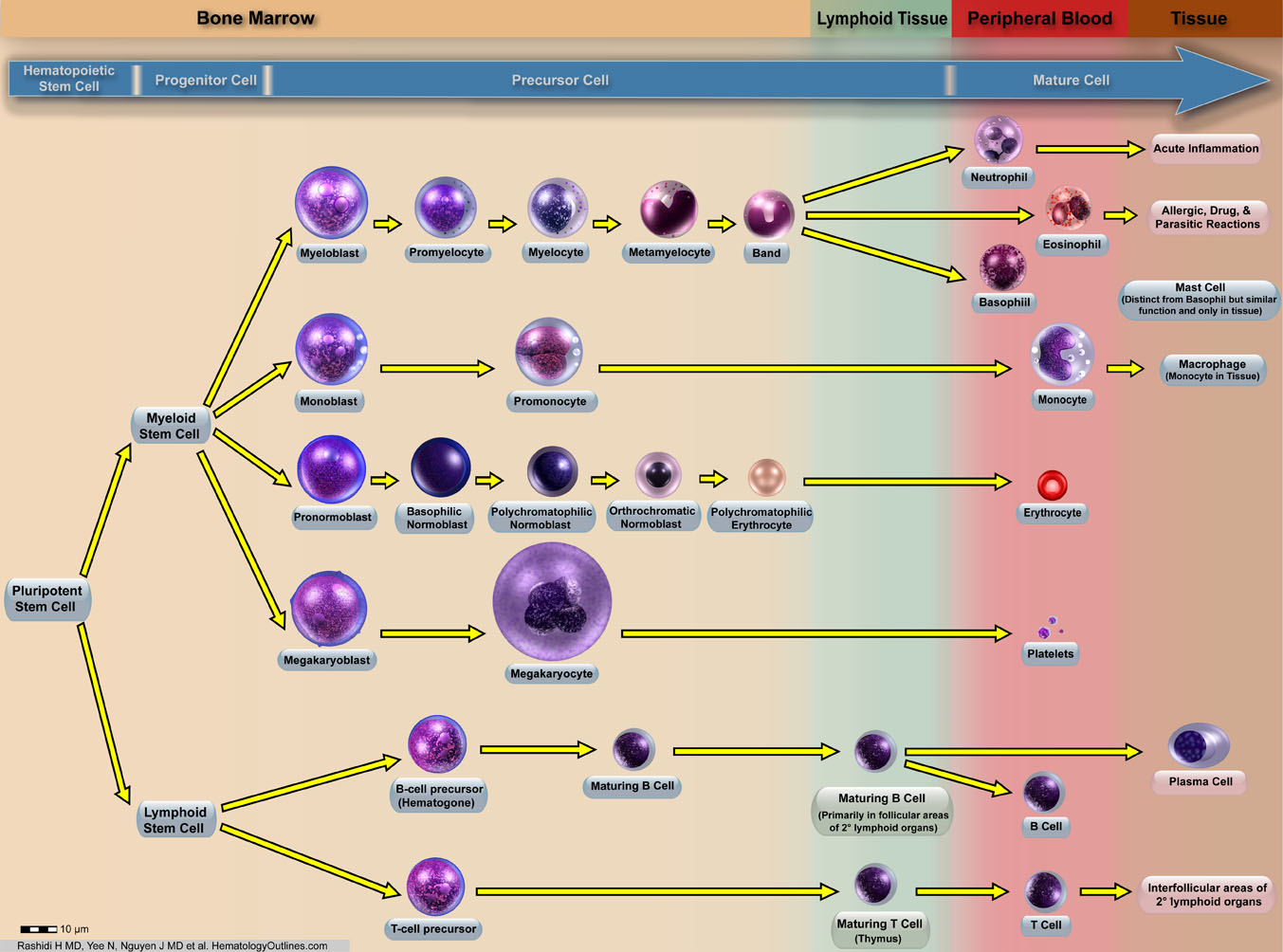
| Hematopoietic cells | These are bone marrow cells that include myeloid (Granulocytic, Monocytic, Erythroid, and megakaryocytic lineages) and lymphoid (T-cell, B-cell, and Nk-cell) cells.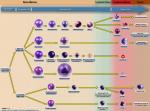 |
|
| Heme | Consists of Iron in the center of a porphyrin Ring. Heme is a constituent of certain oxygen binding molecules such as hemoglobin or Myoglobin and in its normal state its iron is in the ferrous state (Fe++). When oxidized, the Fe++ (Hemoglobin) becomes Fe+++ (methemoglobin). An example of this is noted in bleeding patients when their bright red blood (Fe++) is exposed to outside air and oxidized forming a dark brown color (Fe+++). | |
| Hemoglobin | Abbreviated as Hb. A major protein in Red blood cells (RBCs) that is responsible for carrying oxygen. It is composed of four peptide chains. The three major normal hemoglobins are Hb-A ( 2 alpha chains and 2 Beta chains), Hb-A2 (2 alpha chains and 2 delta chains), and Hb-F (2 alpha chains and 2 gamma chains). The Hemoglobin value can be measured along with Hct, WBC and platelets as part of a routine CBC. Decreased hemoglobin and Hct levels are indicative of anemia. The normal ratio of hemoglobin to hematocrit is about 1:3 (e.g. Hemoglobin of 10 g/dL usually correlates with a Hematocrit of 30%). | |
| Hemoglobin C disease | A hemoglobinopathy that is due to a mutation that changes a hydrophilic residue (glutamate) on the hemoglobin's beta chain to another hydrophilic residue (Lysine). Note: Compared to the sickle cell mutation, the clinical effects are not as significant since the change is from one hydrophilic to another hydrophilic residue versus the change in Sickle cell anemia which involves a hydrophilic residue being replaced by a hydrophobic residue.
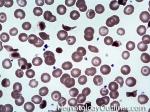 |
|
| Hemoglobin E disease | A hemoglobinopathy that results from a mutation that leads to substitution of Glutamic acid to Lysine (hydrophilic to hydrophilic). Note: Compared to the sickle cell mutation, the clinical effects are not as significant since the change is from one hydrophilic to another hydrophilic residue versus the change in Sickle cell anemia which involves a hydrophilic residue being replaced by a hydrophobic residue. | |
| Hemoglobin H | An Abnormal hemoglobin (Beta chain tetramer) that is the result of excess B-chain production due to marked underproduction of alpha chains in severe alpha thalassemia.
|
|
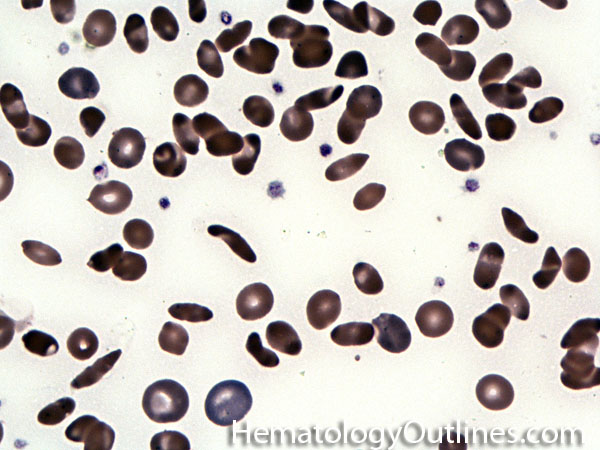
| Hemoglobin S disease | A hemoglobinopathy. Homozygous (Hb-SS) is sickle cell disease (symptomatic) and heterozygous is Sickle cell trait (Usually asymptomatic). hemoglobin S is the result of a mutation that changes the Glutamic acid (hydrophilic) residue on the Beta chain into Valine (hydrophobic residue). The hydrophobic residue repels surrounding water and ultimately induces the deoxy hemoglobin molecules to polymerize, deforming the the red blood cell (sickle shaped).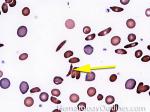 |
|
| Hemoglobinopathy | Genetic diseases that are associated with abnormal hemoglobins due to a mutation that usually leads to a structural abnormality in the hemoglobin molecule. Examples include sickle cell disease (HbSS), HbSC, HbE, and HbCC disease. In contrast to hemoglobinopathies, the defect in thalassemias leads to underproduction of the globin chains rather than a structural abnormality. | |
| Hemolytic anemia | A subtype of anemia that is due to RBC hemolysis. 2 main types: Intravascular (can see schistocytes in PB) & Extravascular (can see spherocytes in PB). Lab findings include a low Hb and Hct, elevated reticulocyte count, increased bilirubin (mainly indirect bili), hemoglobinemia, hemoglobinuria & sometimes hemosiderinuria. Decreased haptoglobin can also be seen and is more prominent in intravascular hemolysis. Causes may be congenital or inherited (e.g. sickle cell disease, hereditary spherocytosis, pyruvate kinase deficiency and G6PD deficiency) or acquired (infection-associated, immune-associated (Coombs positive), hypersplenism-associated, burn injury associated, or mechanical valve associated. Certain aspects of intravascular hemolysis such as schistocytes could also be seen in Microangiopathic Hemolytic Anemias which include DIC, TTP, and HUS. | |
| Hemolytic Uremic Syndrome | AKA HUS is a microangiopathic hemolytic anemia that is characterized by Renal failure (uremia) and thrombocytopenia. Mostly seen in children and preceded by diarrhea caused by E. coli O157:H7. Similar to TTP remember the mneumonic "Brain FART" which is Brain for neurologic problems (more commonly associated with TTP than HUS), F for Fever, A for Anemia, R for Renal failure (more commonly associated with HUS than TTP), and T for Thrombocytopenia. | |
| Hemolytic disease of newborn | Also known as HDN is an alloimmune reaction of maternal IgG molecules (directed against fetal RBC antigens) that cross the placenta and induce hemolysis and destrcution of the fetal RBCs. | |
| Hemophilia | A group of hereditary genetic coagulation disorders that includes Hemophilia A, Hemophilia B, and Hemophilia C. Hemophilia A and B are X-linked disorders while Hemophilia C is an autosomal disorder. Hemophilia A is the most common form of the disorder and is characterized by Factor VIII deficiency. Hemophilia B is less common and is characterized by Factor IX deficiency. Hemophilia C is a lack of Factor XI. Symptoms may vary in severity but usually consist of internal and/or external bleeding (e.g. large joint bleeds). | |
| Hemophilia A | An inherited X-linked bleeding disorder resulting from Factor VIII deficiency. It is the most common form of hemophilia. Symptoms vary depending on the severity and can include hemarthrosis, hematuria, epistaxis, bruising, and prolonged bleeding from cuts or tooth extraction. Typically the patient will have an elevated PTT and normal PT and bleeding time. Serum level of Factor VIII is also decreased. Treatment includes replacement of Factor VIII. | |
| Hemophilia B | An inherited X-linked bleeding disorder resulting from Factor IX deficiency. Less common than hemophilia A. Symptoms are similar to Hemophilia A (see Hemophilia A). The patient will have an elevated PTT and normal PT and bleeding time. Confirmed by low serum levels of Factory IX. Hemophilia B is treated with replacement of Factor IX. | |
| Hemophilia C | A mild form of hemophilia that has an autosomal inheritance pattern characterized by Factor XI deficiency. Symptoms can be similar to Hemophilia A and B with the exception of hemarthrosis, which is a distinguishing feature. The disease is almost exclusively in Jews of Ashkenazi decent. Because the disease is mild, typically no treatment is required. Some cases such as pre-surgical patients, have been treated with FFP or recombinant Factor XI as necessary. | |
| Hemosiderin | A form of iron-complexed to ferritin or other substances. This form of iron is not present in the blood and can be seen in tissue deposits and other sites. Hemosiderin is a major form of iron storage in the marrow and liver. Hemosiderin can increase abnormally in tissues or body fluids after hemorrhage. Hemosiderin is confirmed by stains for iron such as Prussian blue. | |
| Heparin | a.k.a. unfractionated heparin is a widely used anticoagulant that is also naturally occurring in mast cells and basophils. Effective at preventing the formation of blood clots and is generally used for Acute coronary syndrome, atrial fibrillation, deep-vein thrombosis, and etc. Heparin and its low molecular weight derivatives binds to the enzyme antithrombin (AT) that results in its activation which leads to the inactivation of thrombin and other proteases involved in blood clotting, most notably factor Xa. It is monitored by checking the PTT. A very notable and severe complication of unfractionated heparin is heparin-induced thrombocytopenia (HIT). | |
| Hepcidin | A protein that is produced by the liver and acts as a major regulator of iron homeostasis. Hepcidin inhibits iron absorption from the small intestine and inhibits the release of iron from macrophages by binding the iron channel Ferroportin. Hence, when iron stores are low (such as Iron deficiency anemia) Hepcidin levels will decrease enabling iron absoprtion. In contrast, Hepcidin levels will increase in conditions that are not linked to low iron stores (e.g. Anemia of Chronic disease). | |
| Hereditary elliptocytosis | A congenital RBC structural membrane disorder that is usually Autosomal Dominant and leads to increased numbers of RBCs with an elliptical shape (oval shaped). The defect is usually associated with a cytoskeletal scaffold of the RBC membrane. Mutations in the Spectrin genes (Spectrin protein is a structural protein) is the most common finding. Severe cases can lead to a hemolytic anemia. | |
| Hereditary spherocytosis | A congenital RBC structural membrane disorder that is usually Autosomal Dominant and leads to increased numbers of RBCs with spherical shape (round with no central pallor). The defect is usually associated with a cytoskeletal scaffold of the RBC membrane. Mutations in the Spectrin genes (Spectrin protein is a strcutural protein), Ankyrin gene, band 3 or Band 4.2 are the most common findings. Severe cases can lead to a hemolytic anemia. Usually there is an increase in MCHC. Additionally, the cells are more fragile and will give a positive osmotic fragility test result. | |
| Hirudin | Belongs to the class of bivalent direct thrombin inhibitors, which also include Bivalirudin, Lepirudin, and Desirudin. Called bivalent because they bind both the active site and an allosteric site on the thrombin molecule to prevent coagulation. Mainly used to treat heparin-Induced Thrombocytopenia. The drug is cleared by the kidney. | |
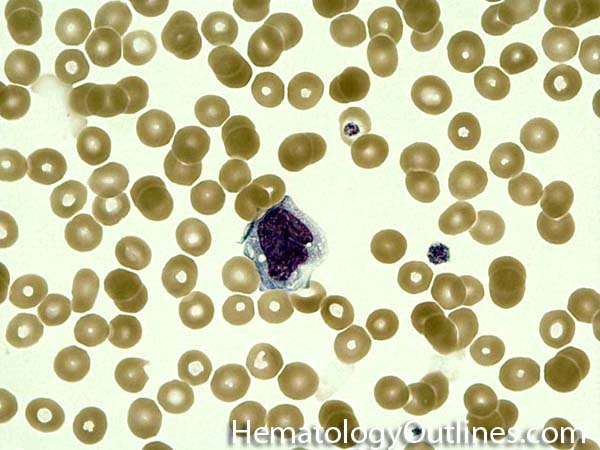
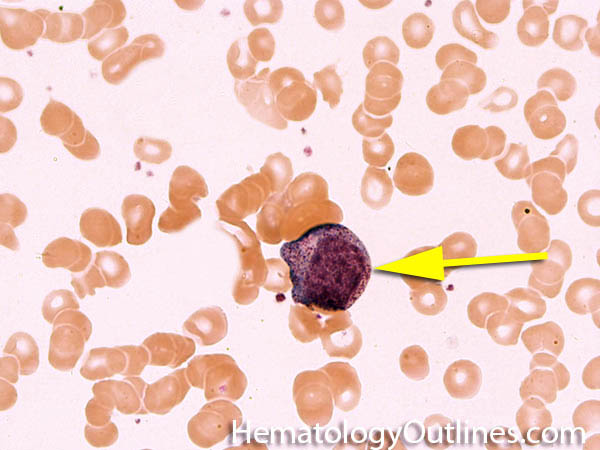
| Histiocytes | This usually refers to tissue macrophages (monocytes that have left the blood and entered tissue). Histiocytes are antigen presenting cells that have phagocytic capabilities. 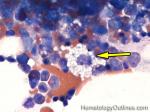 |
|
| Hodgkin lymphoma | An indolent B-cell Lymphoma that usually presents in supradiaphragmatic areas (above the diaphragm such as supraclavicular lymph nodes or Anterior Mediastinum). There is a bimodal age distribution (most patients present in their 20s and 50s). Four main subtypes are: Nodular Sclerosis, Mixed Cellularity, Lymphocyte-rich, and Lymphocyte-depleted. The hallmark of all Classical Hodgkin lymphomas is the Reed-Sternberg cell (RS cell) which usually have the following immunophenotype: dim PAX5+, CD30+, CD15+, CD20-, OCT2- and BOB1-.
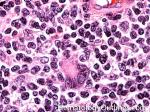   |
|
| Homocysteine | It's the metabolite of the amino acid methionine. Homocysteine may be converted back to methionine with tetrahydrofolate (THF) as a cofactor, and THF is generated with the aid of Vitamin B12. Hence, Hyperhomocyteinemia can be a result of vitamin B12 and/or folate deficiency, and has been linked to cardiovascular diseases. See vitamin B12 and Folate deficiency. | |
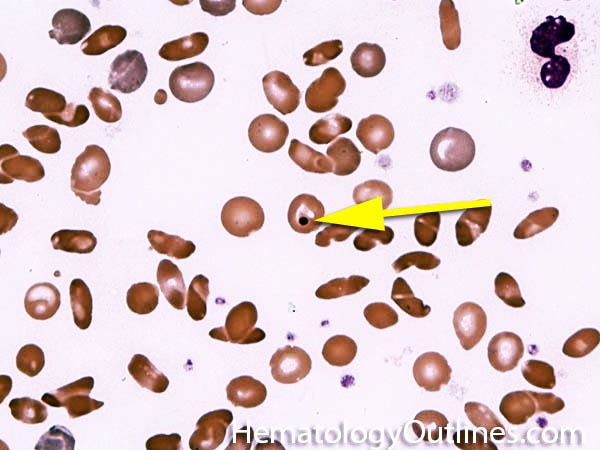
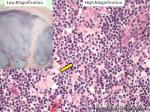
| Howell-Jolly bodies | These are basophilic nuclear remnants (fragments of chromatin/DNA) in RBCs. Mostly seen in post-splenectomy patients or those with asplenia. Commonly seen in adults with sickle cell disease (Hb SS) since these patients have undergone autosplenectomy by the time they reach their adulthood. Remember as opposed to "HEinz" bodies which start with "HE" and stands for hemoglobin, Howell Jolly Bodies don't start with "HE" and are rather DNA or nuclear remnants.
 |
|
| HUS | Hemolytic Uremic Syndrome is a microangiopathic hemolytic anemia that is characterized by Renal failure (uremia) and thrombocytopenia. Mostly seen in children and preceded by diarrhea caused by E. coli O157:H7. Similar to TTP remember the mneumonic "Brain FART" which is Brain for neurologic problems (more commonly associated with TTP than HUS), F for Fever, A for Anemia, R for Renal failure (more commonly associated with HUS than TTP), and T for Thrombocytopenia. | |
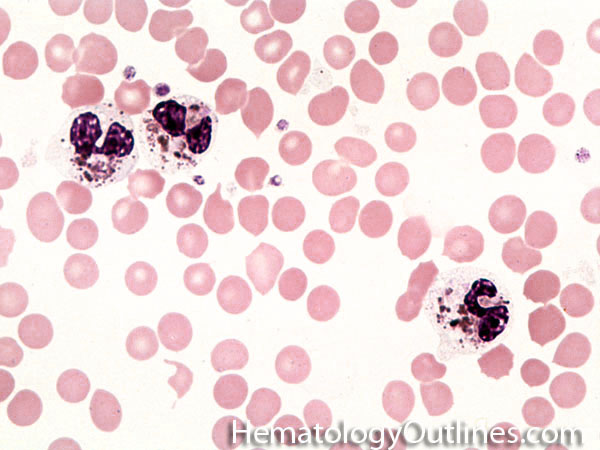
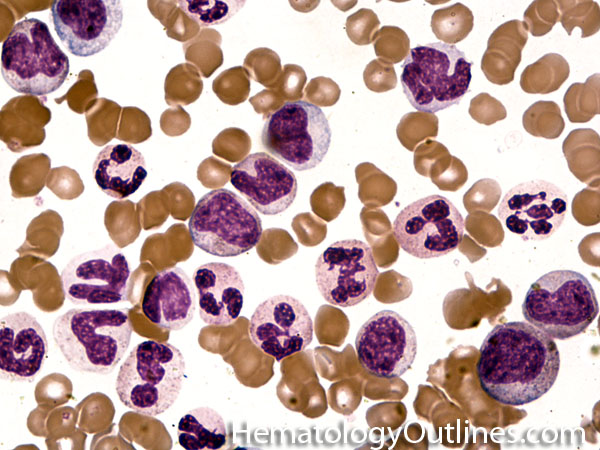
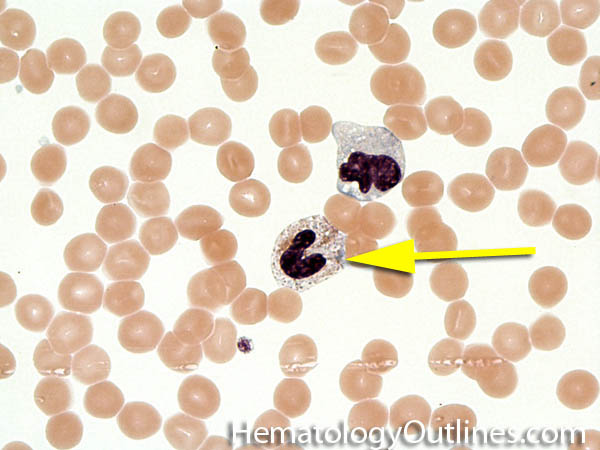
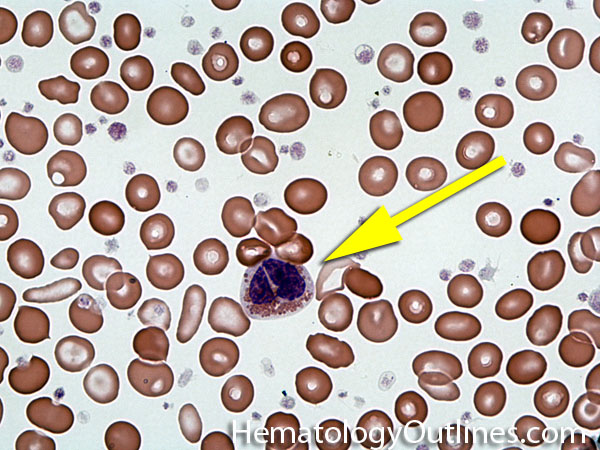
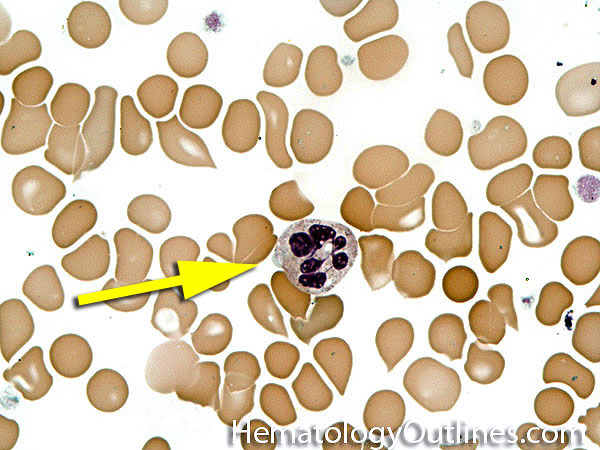
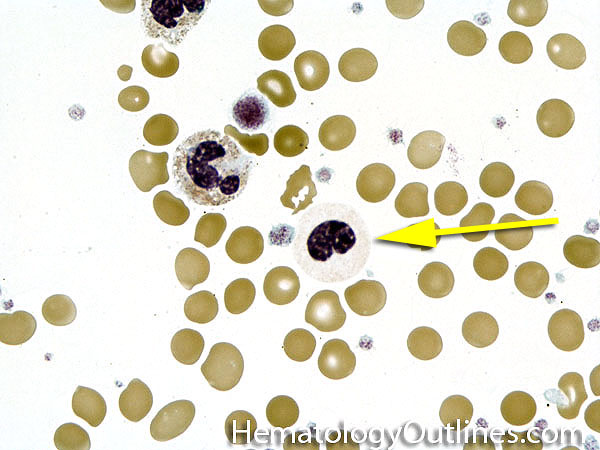
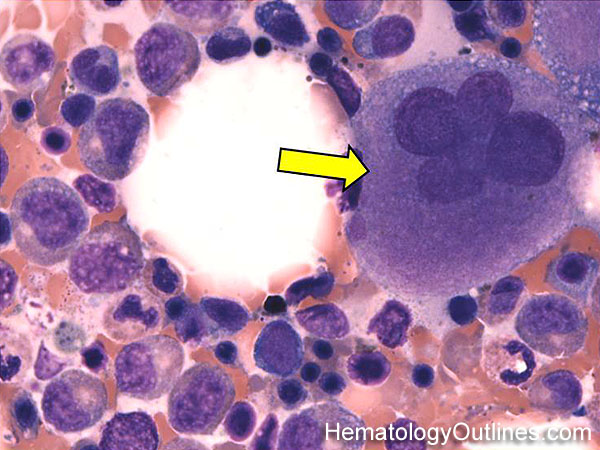
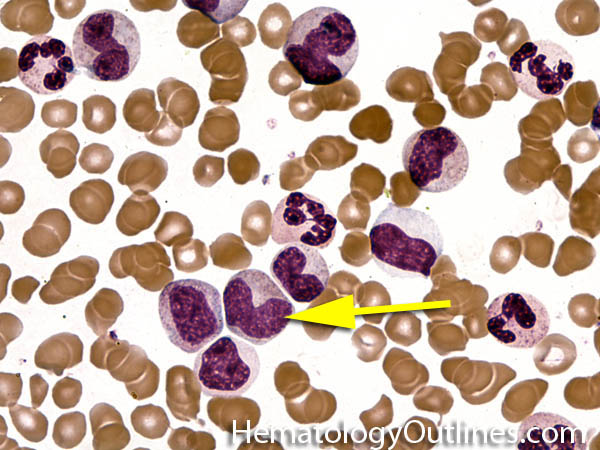
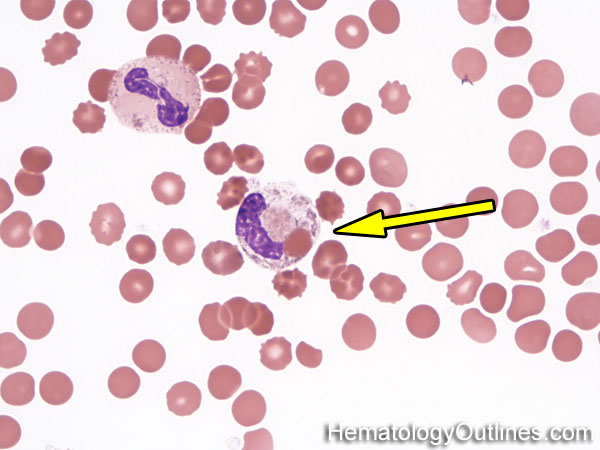
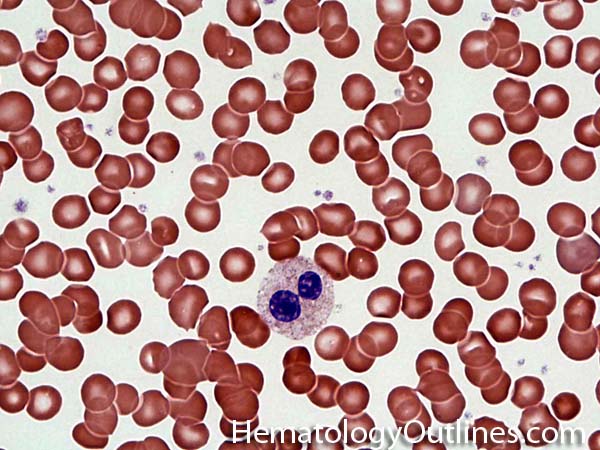
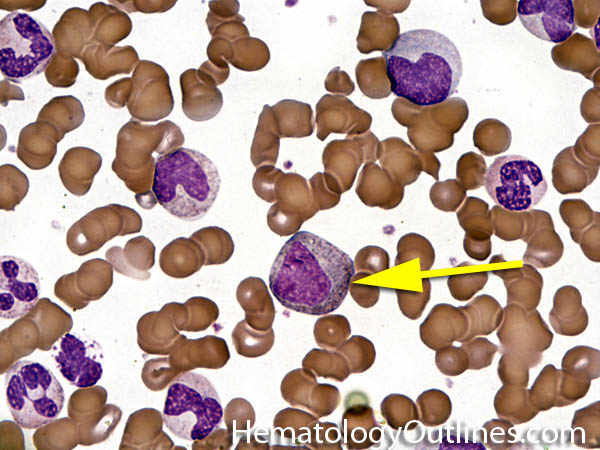
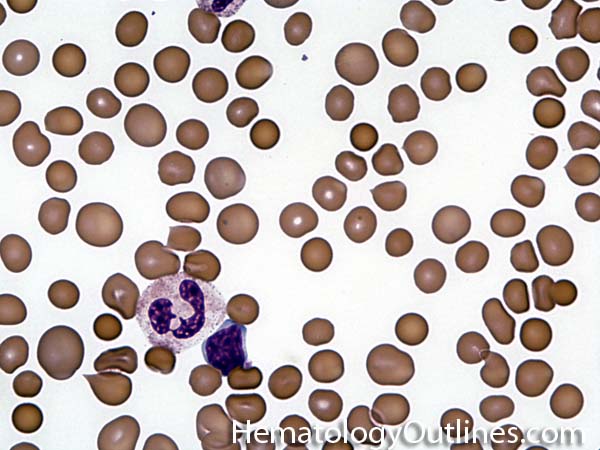
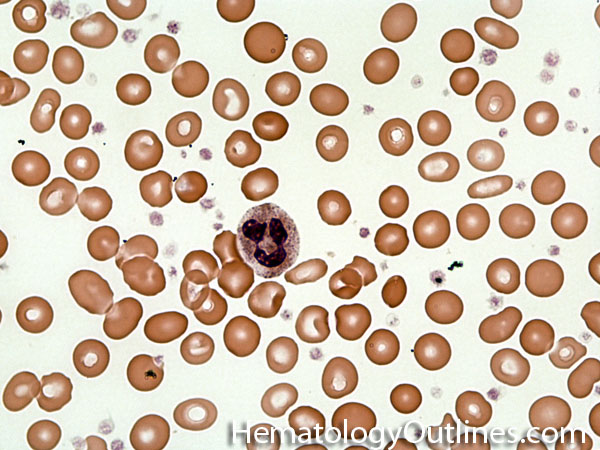
| Hypersegmented neutrophils | These are neutrophils with more than 5 lobes (so 6 or more lobes. Recall, normal neutrophils should have 3-5 lobes). Hypersegmented neutrophils are most commonly associated with B12 or folate deficiency (two common causes of a macrocytic anemia).
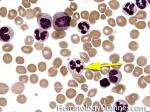 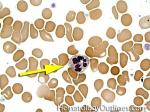 |
|
| Hypogranular | This usually refers to neutrophils and platelets with decreased number of granules. Increased numbers of hypogranular neutrophils (dysplastic neutrophil) may be indicative of an underlying myelodysplastic syndrome.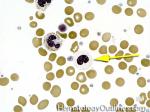 |
|
IBack to top ^ | ||
| IFE | Immunofixation Electrophoresis is an antibody-based method to detect and characterize clonal M-spikes noted on SPEP in patients with suspected plasma cell or B-cell neoplasms. This method reveals the M-spike as clonal with IgG, IgA, or IgM (rarely IgE or IgD) molecules that are either kappa or lambda light chain-restricted. (AKA a clonal antibody from a clonal neoplastic process such as a plasma cell neoplasm or a B-cell Lymphoma). Note: IgG and IgA clones are typically associated with a plasma cell neoplasm (e.g. plasma cell myeloma or MGUS, etc.) while IgM clones may be related to Waldenstrom Macroglobulinemia which is typically associated with lymphoplasmacytic lymphoma. | |
| Imatinib | AKA Gleevec is a tyrosine kinase inhibitor used in treatment of CML by inhibiting the philadelphia chromosome's tyrosine kinase product (ABL1). | |
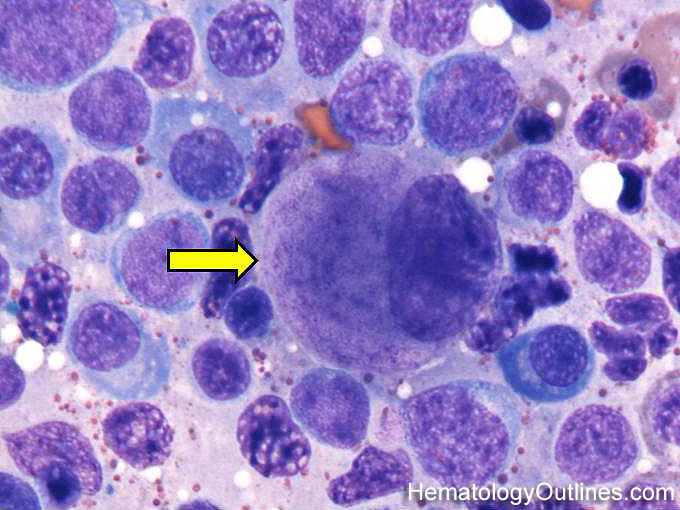
| Immunoblasts | Immunoblasts can be of B-cell or T-cell origin and reside in the perifollicular zones of secondary lymphoid tissue (such as lymph node) where they are morphologically indistinguishable. They are large cells with abundant cytoplasm, large nucleus with open chromatin, and a single centralized large nucleolus. They are part of the developmental stage of lymphocytes which follow the presentation of an antigen by MHC class II positive antigen presenting cells. | |
| Immunoglobulin | The term is sometimes interchangeably used with antibody, a protein that is produced by B cells (in terminally differentiated B-cells known as plasma cells these immunoglobulins are produced and secreted and are not cell surface bound) which recognize and bind to other proteins or substances (foreign or self antigens). Antibodies can be part of an immune response and help eradicate viruses and bacteria or may be part of an autoimmune destructive processes in hematology such as autoimmune hemolytic anemia or immune mediated platelet destruction (e.g. ITP). The different classes of antibodies are usually based on their heavy chains (e.g. IgA, IgG, IgE, IgD, & IgM). | |
| Infectious mononucleosis | A disease that is usually due to Epstein Barr Virus (EBV). More common in younger patients and may present with lymphadenopathy and in severe cases with organomegaly (hepatomegaly and/or splenomegaly). The lymphadenopathy may sometimes mimic a Lymphoma. | |
| INR | a.k.a international normalized ratio is a method to standardize the PT due to the variations between different batches of manufacturer's tissue factor that are used in the PT test. The normal range for INR is 0.8-1.2. Risk of bleeding increases with elevated INRs (see PT. see coumadin.) | |
| International Normalized Ratio | a.k.a INR is a method to standardize the PT due to the variations between different batches of manufacturer's tissue factor that are used in the PT test. The normal range for INR is 0.8-1.2. Risk of bleeding increases with elevated INRs (see PT. see coumadin.) | |
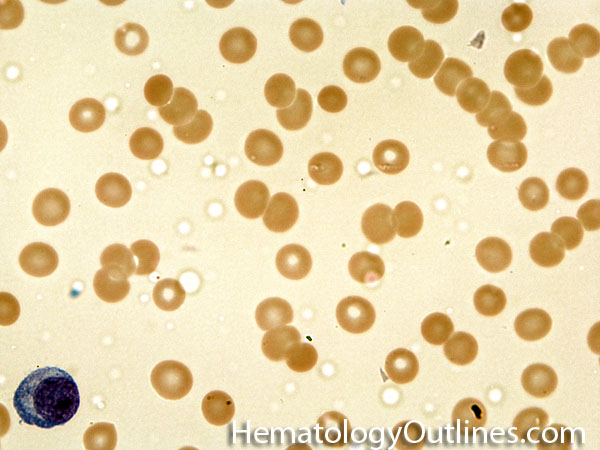
| Iron deficiency anemia | A microcytic anemia (MCV <80) that is due to iron deficiency. Iron deficiency commonly results from Chronic blood loss plus insufficient dietary intake. The blood loss can be either from menstrual bleeding, gastrointestinal bleeding from diverticuli, GI neoplasms, etc. Typically the lab values that correlate with iron deficiency are a low ferritin, saturation index (Fe/TIBC) of <10%, elevated TIBC, and low hepcidin. Additionally, the blood smear shows microcytic hypochromic red cells, ovalocytes called "pencil cells" (sometimes), with anisocytosis, reflected by a high RDW.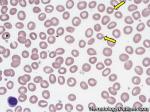 |
|
JBack to top ^ | ||
| JAK2 | Also known as Janus Kinase 2 is a protein whose signaling is involved in many receptor families including but not limited to erythropoietin and thrombopoietin Receptors. Mutation of JAK2 has been identified in many myeloproliferative neoplasms such as polycythemia vera, Essential Thrombocythemia and Primary Myelofibrosis. | |
KBack to top ^ | ||
| Kell | An RBC antigen group that contains immunogenic RBC antigens which may cause transfusion reactions or hemolytic_disease_of_newborn. | |
LBack to top ^ | ||
| LA | a.k.a. Lupus anticoagulant is an immunoglobulin that binds to phospholipids and interferes with the coagulation process in vitro leading to a prolonged PTT. Although the PTT is elevated in LA, the name is a misnomer because it is not an anticoagulant but rather causing thrombosis in vivo. Also most patients with LA do not have Lupus (SLE) . When presented with a prolonged PTT, a mixing study is performed. If the PTT is corrected, then LA is excluded. However, if the clotting time is still prolonged, then the presence of an inhibitor is confirmed, which includes LA. Confirmation of LA is then confirmed through a phospholipid sensitive test such as Dilute Russell’s Viper Venom Time (see dRVVT). | |
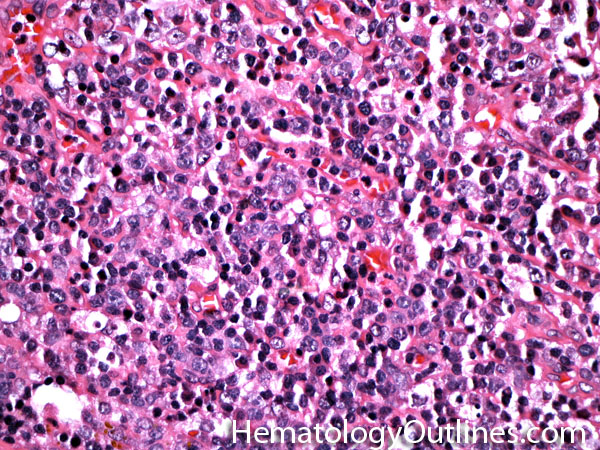
| Large cell lymphoma | Includes Diffuse Large B-cell Lymphoma (DLBCL) and Anaplastic Large Cell Lymphoma (usually a T-cell lymphoma). By definition the neoplastic cells in these lymphomas are typically three times the size or larger than small mature lymphocytes.
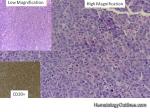 |
|
| Large granular lymphocytes | Also known as LGLs, these are T-lymphocytes with an increased amount of cytoplasm and multiple small red cytoplasmic granules. Increased numbers could be seen in reactive conditions (such as infections) or an indolent neoplastic process such as the T-cell Leukemia known as T-LGL leukemia.
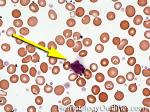 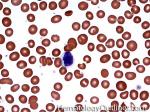 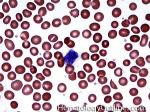 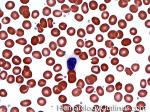 |
|
| Left shifted | Usually refers to seeing less mature cells of a certain lineage. Commonly refers to seeing an increased amount of less mature myeloid (granulocytic) precursors such as myelocytes, metamyelocytes and bands in the blood or bone marrow. | |
| Lenalidomide | Brand name: Revlimid is a chemotherapy drug used to treat plasma cell myeloma and myelodysplastic syndromes (MDS), specifically in patients with deletion 5q with or without additional cytogenetic abnormalities. Side effects include marrow toxicity resulting in neutropenia and thrombocytopenia. | |
| Leukemia | Literally means white blood and refers to a neopastic hematopoietic neoplasm of lymphoid or myeloid origin that involves the peripheral blood. They can be immature (Acute) or mature/maturing (Chronic). Acute leukemias include acute myeloid leukemia (AML) and Acute Lymphoid Leukemia (ALL). Examples of maturing/mature leukemias include chronic myelogenous leukemia (CML) and chronic lymphocytic leukemia (CLL). | |
| Leukocytes | Means "White cells" and refers to the White blood Cells (WBCs) which include granulocytes (Neutrophil, Eosinophil, Basophil), monocytes, and lymphocytes and their precursors. The leukocyte precursors (e.g. promyelocyte, myelocyte, metamyelocyte) are usually seen in the bone marrow and absent in blood of normal individuals. | |
| Leukoerythroblastosis | Refers to the presence of white blood cell precursors and nucleated RBCs in the peripheral blood. This may be due to an underlying marrow infiltrative process, severe infection or severe hemolysis.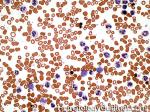 |
|
| Lipocyte | These are fat cells that are typically seen as empty spaces in the bone marrow. The amount of non-fat (cellular) portion of the marrow is the marrow's estimated cellularity. Normal cellularity in people between the age of 15 to 75 is expected to be 100 minus the patient's age. Hence, 70% marrow cellularity in a 30 year old patient is the expected norm (100-30=70%). While 70% cellularity in a 75 year old patient is considered a Hypercellular marrow. | |
| Liver disease coagulopathy | Coagulopathy due to liver disease. Most coagulation factors are made in the liver (majority are made in the liver hepatocytes while factor VIII is made in the reticuloendothelial cells in liver or other sites). Hence, in end-stage liver disease (e.g. cirrhosis) deficiency of these factors usually leads to coagulopathy. | |
| Low molecular weight heparin | Low molecular weight derivative of unfractionated heparin (e.g. enoxaparin, dalteparin, tinzaparin) that prevents coagulation by targeting factor Xa activity rather than antithrombin activity, as in unfractionated heparin. Usually does not require monitoring with PTT (as opposed to unfractionated heparin) and markedly reduces the risk of Heparin-Induced Thrombocytopenia (HIT). Can be monitored with anti-factor Xa assay in certain clinical scenario. | |
| Lupus anticoagulant | a.k.a. LA is an immunoglobulin that binds to phospholipids and interferes with the coagulation process in vitro leading to a prolonged PTT. Although the PTT is elevated in LA, the name is a misnomer because it is not an anticoagulant but rather causing thrombosis in vivo. Also most patients with LA do not have Lupus (SLE) . When presented with a prolonged PTT, a mixing study is performed. If the PTT is corrected, then LA is excluded. However, if the clotting time is still prolonged, then the presence of an inhibitor is confirmed, which includes LA. Confirmation of LA is then confirmed through a phospholipid sensitive test such as Dilute Russell’s Viper Venom Time (see dRVVT). | |
| Lymph node | A peripheral or secondary lymphoid tissue where B-cell migrate to complete their maturation (B-cells undergo somatic hypermutation and class switching of heavy chains in the germinal center of secondary follicles in lymph nodes and other secondary lymphoid tissues such as Mucosa-Associated Lymphoid Tissue and spleen). Mature B-cells could ultimately become plasma cells or memory B-cells in the lymph nodes or other secondary lymphoid tissues. Additionally, the interfollicular areas of these secondary lymphoid tissues are mostly comprised of mature T-cells.
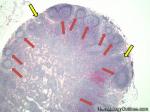 |
|
| Lymphoblast | Immature cell of the lymphoid lineage. In an abnormal neoplastic disorder such as Acute Lymphoblastic Leukemia/Lymphoma (ALL) they are either of a B-cell (B-ALL) or T-cell (T-cell) origin.
(B-lymphoblasts)
(T-lymphoblasts)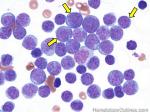 |
|
| Lymphocytes | A type of leukocyte or WBC (second most prevalent WBC in a normal peripheral blood. Most prevalent is neutrophils followed by lymphocytes followed by monocytes, followed by eosinophils and ultimately basophils which are rarely seen). The three main types of lymphocytes include B-cells, T-cells and NK-cells.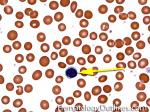  |
|
| Lymphocytosis | This typically refers to an increase in the absolute count of lymphocytes in the peripheral blood. This is also sometimes referred to as "Absolute Lymphocytosis" where the absolute count is usually greater than 4000. Common causes of lymphocytosis are infections (usually viral but sometimes bacterial such as B. pertussis) and B cell neoplasms especially CLL. |
|
| Lymphoma | A neoplastic malignancy of lymphoid origin (B-cell, T-cell or NK cell in origin) that involves some tissue (e.g. lymph node, Spleen, Liver, Stomach, etc.). The T-cell and NK cell lymphomas are typically Non-Hodgkin lymphomas while B-cell lymphomas can be either Non-Hodgkin or Hodgkin lymphomas. | |
| Lymphoma mnemonic | A mnemonic for common translocations associated with several mature B-cell lymphomas. |
|
| Lymphoplasmacytic lymphoma | An indolent B-Lymphoma which is characterized by small-intermediate sized neoplastic lymphocytes and plasma cells. Often associated with Waldenstrom Macrogammaglobulinemia (usually showing an increased amount of clonal IgM by serum protein electrophoresis and Immunofixation Electrophoresis). | |
| Lymphoproliferative disorder | Usually synonymous with Lymphoma. | |
MBack to top ^ | ||
| Macrocytic | Usually refers to Macrocytic (larger) RBCs as seen in macrocytic anemias. Characterized by an elevated MCV (larger RBCs with MCV >100fL). On blood smears, macrocytic red cells are larger than a lymphocyte nucleus. Common causes include B12 or folate deficiency, Liver disease, increased reticulocytes, drug-induced (e.g. methotrexate), and MDS.
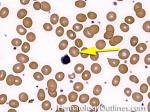 |
|
| Macrophages | Monocytes that have left the blood and entered tissue to serve various functions including phagocytosis of debris and modulate the immune response. 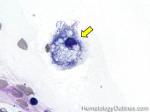 |
|
| Mantle cell lymphoma | An intermediately (between a low grade and high grade Lymphoma) aggressive CD5 positive CD23 negative B-cell lymphoma that is usually due to t(11;14) leading to the overexpression of Cyclin D1 (BCL1). This is in contrast to CLL which is usually CD5+,CD23+ and BCL1 (CyclinD1) negative.
 |
|
| Mast cells | They have many similarities to basophils. Masts cells are usually noted in tissue and bone marrow but absent from peripheral blood (unlike basophils which are in bone marrow and peripheral blood). Mast cells can release substances such as histamine in allergic reactions. Neoplastic mast cells cause cutaneous and systemic mastocytosis. Both normal and abnormal mast cells are typically positive for Mast cell tryptase and CD117 (C-Kit). However, most abnormal mast cells may aberrantly express CD2 and CD25 (T-cell markers).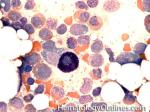 |
|
| May-Hegglin anomaly | Due to Myosin Heavy Chain 9 (MYH-9) mutation. Patients usually show Dohle-like bodies in their neutrophils along with giant platelets in peripheral blood.
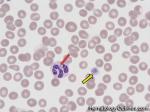 |
|
| MDS | Stands for myelodysplastic syndrome. A heterogeneous group of Chronic myeloid neoplasms which usually share the following features: cytopenia (decrease blood counts such as anemia, thrombocytopenia, or neutropenia) with the exception of MDS with isolated 5q- which may show an increase in platelets), lack of splenomegaly and a hypercellular marrow. The most common MDS include RAEB-I, RAEB-II, RCUD (e.g. RA or RARS), RCMD, and MDS with isolated 5q-. RAEB have the worst prognosis (higher liklihod of transforming into AML) while MDS with isolated 5q- has the best prognosis. | |
| MDS with isolated 5q- | Also known as MDS with isolated 5q deletion is a type of MDS characteristically showing normal or elevated platelet count with a bone marrow displaying increased numbers of hypolobated megakaryocytic along with erythroid hypoplasia. The normal or increased platelet count is unusual amongst MDS, and when the 5q- is the sole cytogenetic abnormality, the prognosis is better than most MDS. These patients are usually treated with lenalidomide. | |
| Megakaryoblast | Immature (blast) form of the megakaryocytic cell lineage (the lineage responsible for making platelets). Increased numbers may be seen in certain AMLs (M7: Acute Megakaryoblastic Leukemia) and AML look-a-likes such as Transient Myelopoiesis which are both more commonly seen in Down Syndrome patients.
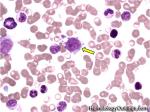 |
|
| Megakaryocytes | Usually the largest cells in the bone marrow that are 10 to 15 times larger than a typical red blood cell. The maturing megakaryocyte grows in size and replicates its DNA without nuclear division resulting in a very large, lobulated, nucleus with many copies of the usual complement of DNA, as much as 64 times most cells. They are multinucleated and responsible for making platelets.
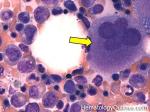 |
|
| Megaloblastic anemia | A cause of macrocytic anemia characterized by large immature RBC precursors (megaloblastoid RBC precursors) in the bone marrow that is the result of impairment in DNA synthesis during RBC production. Common causes include Vitamin B12 deficiency, folate deficiency, chemotherapy-induced (e.g. Methotrexate, 6-Mercaptopurine or Cytosine Arabinoside), and certain malignancies (e.g. MDS). | |
| Megaloblastoid erythroid changes | These are larger than usual erythroid (RBC) precursors with relatively immature nuclear features for the maturity of the cytoplasm (nuclear/ cytoplasmic maturation asynchrony). Causes include certain vitamin deficiencies (e.g. B12 or folate deficiency), drug effects such as methotrexate or due to an underlying myelodysplastic syndrome (MDS).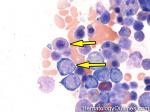 |
|
| Metamyelocyte | A myeloid (granulocytic) precursor that follows the myelocyte and precedes the band in maturation stage of granulocytes. There is indentation of the nucleus but less than 50% of its diameter (as opposed to a band which has an indentation of >50% of the nucleus diameter).
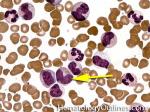 |
|
| Metastasis | Distant spread of a malignancy (e.g. A primary malignancy of lung spreading to the liver or bone, etc.). Note: benign neoplasms usually do not metastasize. | |
| Methemoglobin | Also known as MetHb. The iron in the heme moiety of this hemoglobin molecule is in the Fe+++ state and cannot release the bound oxygen. Note: Normal Hemoglobin's Heme moiety is Fe++ and able to release the bound oxygen | |
| Methylmalonic acid | a.k.a. MMA is a Dicarboxylic acid. When linked to coenzyme A, methylmalonyl-CoA is converted to succinyl-CoA using Vitamin B12 as a cofactor in the Kreb cycle. Vitamin B12 deficiency causes elevated levels of methylmalonic acid (MMA). See vitamin B12 deficiency. | |
| MGUS | Monoclonal Gammopathy of Uncertain Significance are part of plasma cell neoplasms and thought to be precursors to plasma cel myeloma. Similar to the other plasma cell neoplams, they typically display an M-spike on Serum Protein Electrophoresis and shown to be clonal by IFE. | |
| Microangiopathic hemolytic anemia | As the name (microangio) implies, it is a small vessel disease associated hemolytic anemia (intravascular hemolytic anemias). Examples include HUS, TTP, DIC and sometimes due to malignant hypertension. | |
| Microcytic | Usually refers to microcytic (small) RBCs that are seen in microcytic anemia. These are anemias with small RBCs (MCV of <80fL). The Meumonic for Microcytic anemia is "Puny TICS". Both Puny and TICS are small things. Hence, small RBCs with low MCV. P in Puny for Pyridoxine deficiency, T for thalassemia, I for Iron deficiency, C for Chronic disease anemia (sometimes but most are normocytic) and S for sideroblastic anemia (such as in Pb poisoning or congenital sideroblastic anemia).
|
|
| Micromegakaryocytes | These are small abnormal (dysplastic) non-lobated megakaryocytes that are usually associated with an underlying Myeloid neoplasm such as myelodysplastic syndrome.
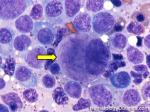 |
|
| Microspherocyte | Small round RBCs with no central pallor that may be seen in in blood smears from patients with immune-mediated hemolytic anemia, burn injuries, and hereditary spherocytosis. | |
| Mixing study (1:1 mix) | A laboratory test used to distinguish factor deficiencies versus factor inhibitors such as lupus anticoagulant. The test is performed by mixing the patient's plasma with a known normal control plasma in a 1:1 ratio. If the 1:1 mixing study results in the correction of PT and/or PTT, then the patient most likely has a factor deficiency. Follow-up factor assays can be performed to further identify the deficiency. No correction mostly likely indicates the presence of a factor inhibitor (most likely an antibody). For patients on anticoagulant therapy, the test should not be performed until the anticoagulant is discontinued. In some situations, the inhibitors are time-dependent, therefore, the 1:1 mix is incubated for 2 hours at 37° C first. An example of such a time-dependent inhibitor is the Factor VIII inhibitor. | |
| MMA | a.k.a. methylmalonic acid is a Dicarboxylic acid. When linked to coenzyme A, methylmalonyl-CoA is converted to succinyl-CoA using Vitamin B12 as a cofactor in the Kreb cycle. Vitamin B12 deficiency causes elevated levels of methylmalonic acid (MMA). See vitamin B12 deficiency. | |
| Monocytes | A type of white blood cell (myeloid lineage). These are usually the largest circulating cells in the blood and usually 2-3 times the size of a mature lymphocyte. Their nucleus is usually folded, and there is a large amount of light blue cytoplasm and commonly show cytoplasmic vacoules. Monocytes that leave the blood and enter tissue are called macrophages.
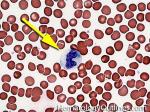 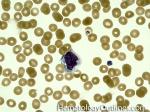 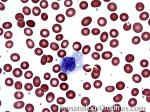 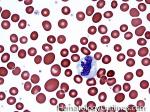 |
|
| Mott cells | These are enlarged plasma cells with multiple cytoplasmic globules which are comprised of immunoglobulins. Mott cells can be seen in certain non-neoplastic (e.g. certain autoimmune diseases) and neoplastic (e.g. plasma cell neoplasms such as plasma cell myeloma, AKA Mutiple Myeloma) processes. | |
| MPN | A group of mature or maturing myeloid neoplasms that usually share the following features: Cytosis refers to the increase in various blood counts (red count, white count, platelet count) with the exception of Primary Myelofibrosis), splenomagly (less common in ET patients), and hypercellular marrow (except in Primary Myelofibrosis or spent phase of the some MPNs). The most common MPNs include CML, PV, ET, and Primary Myelofibrosis. CML has the t(9;22) translocation (BCR/ABL1) while PV, ET and Primary Myelofibrosis usually show the JAK2 mutation. | |
| Multiple myeloma | Also known as plasma cell Myeloma. A plasma cell neoplasm that is usually characterized by increased numbers (10% or more) of monoclonal plasma cells in the bone marrow. Two main types: Asymptomatic (Smoldering) and Symptomatic (characterized by one or more of the following CRAB findings: Remember "CRAB", C for hyperCalcemia, R for Renal disease associated with multiple myeloma, A for anemia associated with the multiple myeloma, and B for lytic Bone lesions).
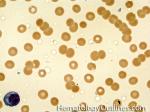 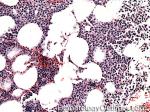 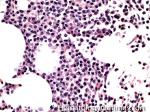 |
|
| Mycoses fungoides | Also known as MF is the most common skin lymphoproliferative disorder. These are usually CD4+ T-cell lymphomas with characteristic epidermotropism (neoplastic cells crawling up into the epidermis) and Pautrier's micro abscesses (collection of neoplastic cells around a dendritic cell in the epidermis) | |
| Myeloblasts | An immatute (blast) form of the myeloid lineage. Normal bone marrow usually has a range of 1-3% myeloblasts. Increased numbers are usually secondary to an underlying myeloid neoplasm such as MDS (RAEB where is there <20% myeloblasts) or AML (which usually has >20% myelobasts).
|
|
| Myelocyte | A granulocytic precursor that follows promyelocyte in the maturation step of granulocytes. Recall maturation steps of granulcytes: myeloblast to promyelocyte to myelocyte to metamyelocyte to band and finally to segmented granulocyte.
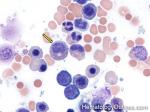 |
|
| Myelodysplastic syndrome | Also known as MDS is a heterogeneous group of Chronic myeloid neoplasms which usually share the following features: cytopenia (decrease in number of a cell line in blood with the exception of MDS with isolated 5q- which may show an increase in platelets), lack of splenomegaly and a Hypercellular marrow. The most common MDS include RAEB-I, RAEB-II, RCUD (e.g. RA or RARS), RCMD, and MDS with isolated 5q-. RAEB have the worst prognosis (higher liklihod of transforming into AML) while MDS with isolated 5q- has the best prognosis. | |
| Myeloproliferative neoplasms | A group of mature or maturing myeloid neoplasms that usually share the following features: Cytosis (increase in a cell line in blood with the exception of Primary Myelofibrosis), Splenomagly (less common in ET patients), and Hypercellular marrow (except in Primary Myelofibrosis or spent phase of the some MPNs). The most common MPNs include CML, PV, ET, and Primary Myelofibrosis. CML has the t(9;22) translocation (BCR/ABL1) while PV, ET and Primary Myelofibrosis many show the JAK2 mutation. | |
NBack to top ^ | ||
| Natural killer cell | Abbreviated as NK-cell. A cytotoxic lymphoid cell that is part of the innate immune system. It's cytotoxicity is due to it's ability of releasing small cytotoxic granules such as perforin and granzymes. They have some similarities to the Cytotoxic CD8+ T-cells. However, unlike the T-cells, NK-cells do not express T-cell Receptors (TCRs), surface CD3 or CD5. They usually express CD56, CD16 and contain some relatively NK specific receptors such as CD94 and KIR (Killer-cell immunoglobulin-like Receptor).
|
|
| Necrosis | A type of cell death. As opposed to apoptosis which can be normal or abnormal, necrotic cell death is almost always abnormal. Common types of necrosis include coagulative necrosis (e.g. tumor necrosis), liquefactive necrosis (e.g. in brain post injury), caseous necrosis (grossly looks cheese-like and may be due to underling tuberculosis or fungal infection), fat necrosis, and hyaline vascular necrosis |
|
| Neoplasm | Usually refers to a clonal growth (tumor). These may be benign or malignant. Benign tumors include adenomas while malignant ones include carcinoma (epithelial), sarcoma (mesenchymal), melanoma (melanocytic), lymohoma and Leukemia (hematopoietic). | |
| Neutrophils | Also known as PMN (for polymorphonuclear), it belongs to the myeloid (granulocytic) cell lineage. Neutrophils usually increase in blood secondary to infection (esp. bacterial). As opposed to eosinophils and basophils, the granules in neutrophils are neutral (light pink) in color.
|
|
| Non-Hodgkin lymphoma | These are mature/maturing neoplastic lymphoid cells (Lymphoma) of either B-cell, NK-cell or T-cell lineage. Examples include Diffuse Large B-Cell Lymphoma (DLBCL) and follicular_lymphoma which are of B-cell origin while Anaplastic Large Cell Lymhphoma (ALCL) and Peripheral T-Cell Lymphoma Not Otherwise Specified (PTCL-NOS) are of T-cell origin. | |
| Normoblast | An Erythroid precursor (nucleated form of Erythroid). | |
| Normochromic/normocytic erythrocytes | These are normal appearing mature RBCs and can be seen in normal individuals or those with certain anemias. As opposed to the immature Erythroid precursosrs which are nucleated, the mature RBCs lack a nucleus and have a central pallor that is usually 1/3 of it's diameter. The main function of the RBC is to carry oxygen through it's hemoglobin molecules.
|
|
| Normocytic | This refers to "normal size" mature RBCs or anemias with normocytic appearance (MCV 80-100 which is the normal range). These anemias include but are not limited to Hemolytic anemia, sickle cell anemia, and Anemia of Chronic disease. | |
| Nucleated red blood cells | These are also known as NRBC which are red blood cells who as the name implies have a nucleus (as opposed to normal mature RBCs that do not have a nucleus). Increased numbers of these less mature erythroid cells can be associated with an underlying infiltrative marrow process, leukoerythroblastic process and sometimes as a response to severe hemolysis.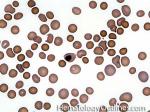  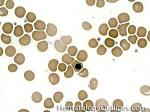 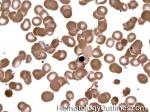 |
|
OBack to top ^ | ||
| Osteoblasts | Resident cells in the bone that are responsible for bone formation. May sometimes resemble plasma cells (as opposed to plasma cells which have a perinuclear cytoplasmic clearing, the cytoplasmic clearing in osteoblasts is away from the nucleus and more in the center of the cytoplasm).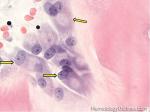 |
|
| Osteoclasts | Multinucleated resident cells in the bone that are responsible for bone resorption. May sometimes resemble other multinucleated cells in the bone marrow such as megakaryocytes (The nuclei of megakaryocytes are connected to each other while the nuclei of osteoclasts appear round and disconnected from one another) | |
| Ovalocytes | Also sometimes referred to as elliptocytes are RBCs with an elongated or oval shape and may be secondary to hereditary elliptocytosis or iron deficiency anemia to name a few.
|
|
PBack to top ^ | ||
| Pappenheimer bodies | Dense, blue-purple granules of iron found in the periphery of the cytoplasm of an RBC. Most commonly, they are associated with splenectomy, either surgical or as a complication of sickle cell anemia. They also can be seen in smears from patients with sideroblastic anemia, and hemolytic anemias. In contrast to Howell-Jolly (HJ) bodies that are a single, very round red cell inclusions, Pappenheimer bodies are smaller, more irregular in outline, and often are multiple per red cell.
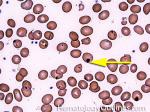 |
|
| Paroxysmal cold hemoglobinuria | This a rare autoimmune hemolytic anemia that is due to cold reacting IgG antibodies to the RBC P antigen. These antibodies bind RBCs at cold temperature and with elevated temperature the bound red cells undergo a complement induced destruction (hemolysis). These biphasic unusual cold reacting IgGs are also known as Donath-Landsteiner antibodies. These patients may undergo hemolysis when exposed to cold temperature and present with hemoglobinuria. Peripheral blood may show some neutrophils with erythrophagocytosis.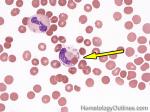 |
|
| Paroxysmal Nocturnal hemoglobinuria | Also know as PNH is an acquired complement-induced hemolytic anemia that is due to a mutation of phosphatidylinositol glycan A (PIGA) gene (on chromosone X) which is resposible for making the cell membrane protein anchoring molecule glycosylphosphatidylinositol (GPI). Notably the gene is on chromosome X but unlike X-linked germline mutations (e.g. hemophilia A) which almost exclusively involve males, this acquired mutation involves males and females equally. The diagnosis is made by flow cytometry and showing loss of CD55 (Complement decay accelerating factor) and CD59 (protectin which usually inhibits the complement membrane attack complex) on multiple blood cell types (RBCs and WBCs). These patients are also at an increased risk of thrombosis. | |
| Pelger-Huet anomaly | An rare inherited condition due to lamin B receptor mutation in which most of the peripheral blood neutrophils present with bilobed nuclei connected with a thin filament of chromatin (similar to the bilobed nuclei noted on normal mature eosinophils but without the eosinophilic granules). In heterozygous individuals these neutrophils appear to maintain their function, and most of these individuals are asymptomatic (hence the term anomaly rather than disease).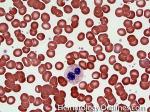 |
|
| Plasma cell | This is a terminally differentiated B-cell that produces and secretes immunoglobulins (antibodies). Some B-cells in the peripheral lymphoid tissues (e.g. lymph node) further differentiate and become plasma cells. Normally plasma cells are not present in the peripheral blood and reside in secondary lymphoid tissues and other tissues such as the bone marrow. It has an eccentric nucleus with a "clock-face" chromatin pattern and abundant blue cytoplasm and a perinuclear clearing.
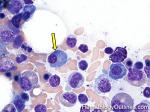 |
|
| Plasma cell Leukemia | This is a very aggressive plasma cell neoplasm with an increased number circulating plasma cells in the peripheral blood (usually >20% of WBCs). | |
| Plasma cell myeloma | Also known as multiple myeloma is a plasma cell neoplasm that is usually characterized by increased numbers (10% or more) of monoclonal plasma cells in the bone marrow. Two main types: Asymptomatic (Smoldering) and Symptomatic (characterized by one or more of the following CRAB findings: Remember "CRAB", C for hyperCalcemia, R for Renal disease associated with multiple myeloma, A for anemia associated with the multiple myeloma, and B for lytic Bone lesions).
|
|
| Plasmacytoma | A plasma cell neoplasm that consists almost entirely of neoplastic plasma cells involving tissue outside of the bone marrow. | |
| Plasminogen | Plasminogen is a zymogen produced by the liver and released into the circulation. Plasminogen is cleaved by enzymes such as tissue-plasminogen activator (t-PA) to form plasmin. Plasmin (a serine protease) is then responsible for degradation of fibrin clot, a process known as fibrinolysis. | |
| Platelet granules | Platelet granules are present within platelets and released during platelet activation. There are three types of granules present within platelets: alpha granules, Delta (dense) granules and Lysosomes. Alpha granules contain the larger molecules such as Platelet Factor 4 (PF4), Platelet-derived growth factor (PDGF), fibrinogen and some other clotting factors. The Delta (Dense) Granules contain the smaller molecules such as Serotonin, ADP, & Calcium. Mnemonic is Alpha stands for the "Bigger" molecules such as proteins and peptides, while Delta or Dense stands for "Diminutive" (tiny) molecules. | |
| Platelets | Cellular fragments (2-3 um in diameter which correlates to about 1/3 to 1/2 the size of a RBC) released from megakaryocytes into the blood and involved in hemostasis.
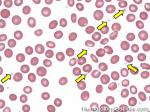 |
|
| Pleuripotential hematopoietic stem cells | These stem cells are derived from totipotent stem cells and can differentiate into any of the hematopoetic cell lines (myeloid or lymphoid). | |
| PNH | Paroxysmal Nocturnal hemoglobinuria is an acquired complement-induced hemolytic anemia that is due to a mutation of phosphatidylinositol glycan A (PIGA) gene (on chromosone X) which is resposible for making the cell membrane protein anchoring molecule glycosylphosphatidylinositol (GPI). Notably the gene is on chromosome X but unlike X-linked germline mutations (e.g. hemophilia A) which almost exclusively involve males, this acquired mutation involves males and females equally. The diagnosis is made by flow cytometry and showing loss of CD55 (Complement decay accelerating factor) and CD59 (protectin which usually inhibits the complement membrane attack complex) on multiple blood cell types (RBCs and WBCs). These patients are also at an increased risk of thrombosis. | |
| Poikilocytosis | The presence in the blood of erythrocytes showing abnormal variation in "shape". Mnemonic is when compared to anisocytosis. Both the words "Shape" and "Poikilocytosis" contain a "P". Therefore Poikilocytosis has to do with RBC Shape variation while Anisocytosis (No "P" in the word) has to do with variation of "size" (No "P" in the word).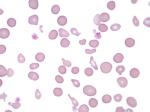 |
|
| Polychromatophilic | Usually refers to the polychromatophilic RBC which is less mature non-nucleated red blood cell with a light basophilic (light blue-purple) color on peripheral smear which is due to the presence of cytoplasmic RNA. Increased numbers may be seen in certain anemias such as hemolytic anemia in which earlier RBCs are being released by the reactive bone marrow in response to the anemia with the earlier release of these less mature RBCs into circulation. Special stain can highlight the RNA molecules in these cells and on the special stain these cell are known as reticulocytes.
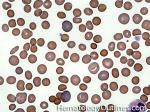 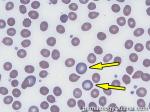 |
|
| Polycythemia vera | A myeloprolifeartive neoplasm that usually carries the JAK2 mutation. Patients with this neoplasm typically present with splenomegaly, and some "cytosis" which is usually in the form of an elevated hematrocrit/hemoglobin with low EPO levels. The bone marrow is usually hypercellular similar to many of the other myeloproliferative neoplasms.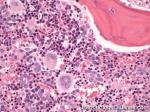 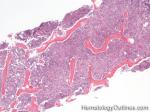 |
|
| Proerythroblast | The earliest erythrocyte precursor in the erythrocyte series which usually contains a large nucleus with several nucleoli and high Nucleus/Cytoplasm ratio (meaning very little cytoplasm).
|
|
| Promyelocytes | A granulocytic precursor that follows myeloblasts and preceeds myelocytes. They usually contain one or more nuceloli and a few or many coarse, reddish-purple, azurophilic (primary) granules.
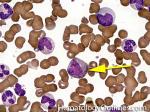 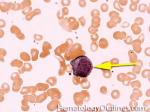 |
|
| Protein C | Protein C is an inactive enzyme form (zymogen) of activated protein C (APC) which plays an important role in anticoagulation. Protein C is a Vitamin K dependent protein produced by the liver. Protein C is activated by thrombin, with protein s as a cofactor, to from APC, a serine protease. APC then acts to inhibit the activation of Factor V and VIII. Protein C deficiency ranges from mild to severe. Depending on the type of deficiency (heterozygous or homozygous), it can lead to an increased risk of thrombosis. | |
| Protein S | Protein S is a Vitamin K dependent glycoprotein produced in the endothelium. It is important in anticoagulation because it acts as a cofactor in the activation of Protein C (see Protein C). Protein S deficiency, caused by a mutation in the PROS1 gene, is rare but can lead to an increased risk of thrombosis. | |
| Prothrombin Time | a.k.a PT is a laboratory test used to measure the integrity of the extrinsic coagulation pathway and commonly used to monitor warfarin (coumadin) therapy. Normal range: 10-13 seconds. It is sensitive to detecting deficiencies in the extrinsic pathway such as factor VII and common pathway factors I, II, V, and X (Note: Common pathway coagulation factors effect both PT and PTT). The test is performed by taking citrated plasma to clot at 37° C after the addition of calcium. tissue factor is then added and the time it takes to clot is measured. The resultant PT is normalized with the international normalized ratio (INR) (see INR). PT is typically increased in Coumadin therapy, Vitamin K deficiency, liver disease, DIC, and deficiencies in extrinsic or common pathway coagulation factor(s). | |
| Pseudo-Gaucher cells | Pseudo-Gaucher cells are histiocytes with cytoplasmic needle-like inclusions, resembling histiocytes noted in Gaucher disease (Gaucher cells). These pseudo-Gaucher cells have been shown in a variety of diseases including but not limited to CML, multiple myeloma, and ALL. | |
| Pseudothrombocytopenia | Pseudo-thrombocytopenia is an artifactual clumping of platelets that can occur after exposure to EDTA. The clumping occurs only in the test tube with EDTA and may be associated with an antibody that reacts to platelet antigens only in the presence of EDTA. Citrated blood can be used to get an accurate platelet count when EDTA causes this problem. | |
| PT | a.k.a prothrombin time is a laboratory test used to measure the integrity of the extrinsic coagulation pathway and commonly used to monitor warfarin (coumadin) therapy. Normal range: 10-13 seconds. It is sensitive to detecting deficiencies in the extrinsic pathway such as factor VII and common pathway factors I, II, V, and X (Note: Common pathway coagulation factors effect both PT and PTT). The test is performed by taking citrated plasma to clot at 37° C after the addition of calcium. tissue factor is then added and the time it takes to clot is measured. The resultant PT is normalized with the international normalized ratio (INR) (see INR). PT is typically increased in Coumadin therapy, Vitamin K deficiency, liver disease, DIC, and deficiencies in extrinsic or common pathway coagulation factor(s). | |
| PTT | Activated Partial Thromboplastin Time is laboratory test that measures the integrity of the intrinsic pathway of coagulation and commonly used for monitoring heparin therapy. It is sensitive to detecting deficiencies in factors involved in the intrinsic pathway such as factor VIII, IX, and XI, common pathway such as factor X or phospholipid-dependent inhibitors such as lupus anticoagulants. As noted above, it is also prolonged if certain anticoagulants such as heparin are present. The test is performed by taking citrated plasma to clot at 37° C after the addition of calcium (a set amount of phospholipid is also added but no tissue factor is added so it will not evaluate the extrinsic pathway). PTT could also be used comparatively in mixing studies when one suspects factor deficiency or presence of inhibitors. In a patient with prolonged PTT (& normal PT), first the presence of heparin must be excluded. Once heparin is excluded, the patient's plasma can be mixed with a known normal plasma sample to see if the PTT corrects. Correction of the PTT in such setting is suggestive of a factor deficiency while lack of correction may imply presence of an inhibitor (antibody), such as lupus anticoagulant. | |
QBack to top ^ | ||
| Q-- | - | |
RBack to top ^ | ||
| RBC | Also called Red blood Cell or erythrocyte, are the most common type of blood cell in the body. It's role is to deliver oxygen via the hemoglobin molecule to various tissues and organs.
|
|
| Reactive lymphocytes | They used to be called atypical lymphocytes. These large lymphocytes may be increased in the peripheral blood smear in certain inflammatory conditions such as an underlying viral infection (e.g. EBV induced mononucleosis, the lymphocytes look so large that they start resembling monocytes). They usually have increased amount of cytoplasm which wraps or hugs the surrounding RBCs. Reactive lymphocytes are typically CD8+ T-cells.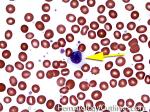 |
|
| Red blood cell | Also called RBC or erythrocyte, are the most common type of blood cell in the body. It's role is to deliver oxygen via the hemoglobin molecule to various tissues and organs.
|
|
| Refractory anemia | A subtype of MDS under the category of Refractory Cytopenia with Unilineage Dysplasia. | |
| Reticulin fibrosis | A type of fibrosis (Reticulin usually refers to type III collagen) made evident by special reticulin stains. Marrows may show increased reticulin for various reasons including metastatic tumors, Lymphoma, and clonal disorders of the marrow (MDS, AML, MPN). Extensive increases in reticulin are characteristic of primary myelofibrosis and a common complication of other MPN. | |
| Reticulocytes | Reticulocytes are immature RBCs that do no contain nuclei but have increased amounts of RNA. On normal peripheral blood smears stained with Wright-Giemsa they appear as polychromatophilic cells (light purple/blue cells are the polychromatophilic cells versus the more mature RBCs stain pink/red) and with special stains (such as supravital stain) one could highlight the excess RNA within the cytoplasm of these RBCs known as reticulocytes. Normal range for reticulocytes in the peripheral blood is 0.5-1.5%. Increased reticulocytes can be seen in the setting of anemias that are due to desctruction of RBCs (such as in autoimmune hemolytic anemia) which causes the marrow to increase production of newer RBCs. Low recticulocytes are usually indicative of marrow underproduction and can be caused by chemotherapy, aplastic anemia, pernicious anemia, bone marrow malignancies, vitamin and mineral deficiencies, Chronic diseases, or lack of erythropoietin production. |
|
| Reticuloendothelial cells | These cells are part of the immune system that comprises certain phagocytic cells such as monocytes and macrophages within the reticular connective tissue as noted in the spleen, liver and the lymph node. | |
| Ring Sideroblasts | Abnormal nucleated RBCs with granules of iron accumulated in perinuclear mitochondria, which can be seen by microscopy after special iron stains such as Prussian Blue. Increased ringed sideroblasts are a diagnostic feature of certain disease states such as the myelodysplastic syndrome Refractory anemia with Ring Sideroblasts. Other causes of ringed sideroblasts include copper deficiency, and zinc toxicity (which could induce copper deficiency). Ring sideroblasts are specifically defined as sideroblasts encircled by 5 or more iron granules and.surrounding at least 1/3 of the nucleus (hence the term " Ring Sideroblasts").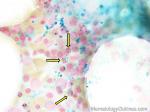 |
|
| Rouleaux formation (rouleaux RBCs) | Stacking of RBCs (like a stack of coins) which is usually due to an increase in the relatively positively charged blood proteins, such as immunoglobulins and fibrinogen which ultimately lead to a decrease in the zeta_potential between the RBCs and ultimately increase the chance of RBCs sticking to eachother. Rouleaux formation can be seen in multiple myeloma, and certain inflammatory conditions.
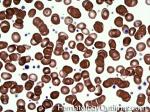 |
|
| Russell bodies | Russell bodies are immunoglobin containing inclusions found in cytoplasm of plasma cells. They can be seen in multiple myeloma but they can also be seen in reactive non-neoplastic plasma cells.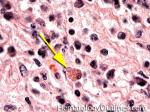 |
|
| Russell Viper Venom Time | a.k.a. Dilute Russell Viper Venom Time (dRVVT) is an in vitro qualitative test for lupus anticoagulant (LA) and usually follows an elevated PTT when LA is suspected. The test is derived from the venom of Russell viper, a power thrombotic agent in vitro. A mixing study is performed combining Russell's viper venom, patient’s plasma, and phospholipids, which is required for coagulation. The presence of lupus anticoagulant would prevent clotting. A prolonged clotting time would be followed up with a confirmatory test where excess phospholipids are added to the mixing study, which should overcome LA and induce clotting. A ratio between clotting time without excess phospholipids and with excess phospholids is then calculated. An elevated ratio is considered positive and consistent with LA. | |
SBack to top ^ | ||
| Schistocytes | These are irregularly shaped assymetrical fragmented RBCs that may have several morphologic forms (e.g. Helmet cells, etc.). It is usually the result of mechanical disruption of the RBCs and many of which are generated by getting stuck to fibrin strands within the vessel wall (intravascular destruction) which may be secondary to a Microangiopathic hemolytic anemia such as HUS, TTP and DIC or a fibrin-independent destructive path such as being broken up by a mechanical heart valve.
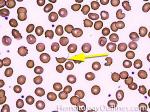 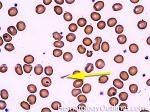 |
|
| Segmented neutrophils | A segmented neutrophil is also referred to as a polymorphonuclear neutrophil (PMN). In a normal CBC, they are the most abundant white blood cells. They are also the first responders recruited to the site of injury such as inflammation which may be secondary to a bacterial infection, environmental exposure, and some cancers.
|
|
| Sezary syndrome | Sezary syndrome is defined as a constellation of 1) erythroderma (with involvement by a cutaneous T-cell Lymphoma which is usually comprised of CD4(+) T-cells), 2) circulating abnormal T-cells in the blood (sezary cells) and 3) lymphadenopathy. On peripheral blood smear these Sezary cells are the abnormal lymphocytes with nuclear membrane irregularities and scant cytoplasm. When compared to the most common cutaneous T-cell lymphoma (mycoses_fungoides), sezary syndrome patients have a much worse prognosis.
|
|
| Sickle cell | Abnormal RBCs seen in sickle_cell_anemia, with rigid sickle shape and reduced flexibility. They have a banana shaped appearance. See Sickle_cell_anemia.
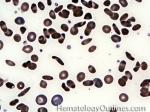 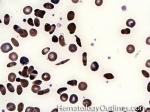 |
|
| Sickle cell anemia | A hemoglobinopathy that is due to homozygosity from a point mutation in the beta-globin gene, where ultimately the glutamic acid (a hydrophilic residue) on position 6 of the beta globin chain is replaced with valine (a hydrophobic residue). The hydrophobic residues are not well suited for the surrounding hydrophilic environment inside the RBC and ultimately lead to aggregation of these abnormal hemoglobin molecules . These "polymerized" hemoglobin molecules cause abnormal RBCs with rigid and sickled shapes. Sickle cells lack the flexibility of normal RBCs and can be trapped within capillaries causing multiple complications (e.g. "sickle cell crisis" which includes vaso-occlusive crisis, hemolytic crisis, aplastic crisis, and sequestration crisis). Sickle cell patients commonly undergo autosplenetcomy during childhood due to infarction of the spleen with subsequent increased risk for developing infections due to encapsulated bacteria such as Streptococcus and H. influenzae, therefore requiring vaccinations. Various other clinical complications include stroke, avascular necrosis, osteomyelitis (Salmonella infection), Chronic pain, pulmonary hypertension, chronic renal failure, and etc. |
|
| Sideroblastic anemia | A disease in which the bone marrow produces sideroblasts rather than normal RBC precursors. It can be caused by a genetic disorder or acquired such as in lead poisoning, zinc excess or sometimes seen with some myelodysplastic syndromes. In sideroblasic anemia, the body cannot properly utilize and incorporate iron into hemoglobin, therefore leading to an excess iron accumulation. The MCV is usually microcytic in sideroblastic anemia due to lead poisoning or congenital sideroblastic anemia while those associated with MDS can be macrocytic. | |
| Sideroblasts | These are nucleated RBCs with a perinuclear deposition of iron granules. A type of these sideroblasts is called Ringed_sideroblasts which can be seen in certain MDS such as Refractory anemia with Ringed_Sideroblasts. | |
| SLL | Small Lymphocytic Lymphoma is an indolent B-cell lymphoma and in simple terms Chronic lymphocytic Leukemia (CLL) involving tissue such as lymph nodes (hence the term lymphoma) . The characteristic immunophenotype is similar to CLL showing dim CD20 (+), CD19 (+), CD5 (+), CD23 (+), CD10 (-), and monotypic dim surface kappa or lambda light chain restricted neoplastic B-cells.
  |
|
| Small lymphocytic lymphoma | Also known as SLL is an indolent B-cell Lymphoma and in simple terms Chronic lymphocytic Leukemia (CLL) involving tissue such as lymph nodes (hence the term lymphoma) . The characteristic immunophenotype is similar to CLL showing dim CD20 (+), CD19 (+), CD5 (+), CD23 (+), CD10 (-), and monotypic dim surface kappa or lambda light chain restricted neoplastic B-cells.
|
|
| Smudge cells | Fragile lymphocytes that are broken down during blood smear preparation. Smudge cells are commonly seen in CLL.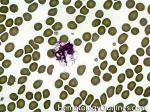 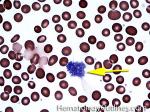 |
|
| SPEP | Serum Protein Electrophoresis is a method that is used to separate and quantitate the different protein components of blood. One of these include the immunoglobulins, and when an M-spike (Monoclonal immunoglobulin band) is seen in this region on the gel, the amount of this potential clonal M-spike is quantitated by this method. Further charcaterization of this clone is done by IFE. | |
| Spherocytes | Sphere shaped erythrocytes (RBCs) with reduced membrane and diameter which can be due to hereditary spherocytosis or autoimmune hemolytic anemia when increased numbers are noted in blood.
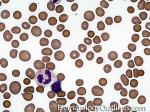  |
|
| Splenic marginal zone lymphoma | A low-grade mature B-cell Lymphoma that usually involves the spleen, peripheral blood and sometimes other organs such as the bone marrow. Characteristic features are splenomegaly (involving the white pulp of spleen as opposed to hairy cell Leukemia which involves the splenic red pulp), moderate lymphocytosis with villous morphology (seen in the peripheral blood smear), and intrasinusoidal pattern of involvement of various organs, especially the bone marrow. Characteristic immunophenotype is similar to other marginal zone lymphomas (triple negative phenotype referring to being negative for CD5, CD10 and CD23).
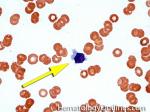 |
|
| Splenomegaly | Splenomegaly is an enlargement of the spleen. Common causes of splenomegaly include infectious mononucleosis, end stage liver disease, certain hemoglobinopathies (e.g. HbSC disease), certain hematological malignancies (Many Myeloproliferitive Neoplasms, some lymphomas/leukemias such as Splenic Marginal Zone Lymphoma and hairy cell Leukemia), infiltration by cancer, and certain metabolic diseases. | |
| Spur cell RBC | Also known as acanthocyte is a RBC with irregular thorn-like projections. Looks like "Cowboy Boot Spurs". These RBCs may be associated with many conditions including but not limited to abetalipoproteinemia, liver disease, malnutrition, asplenia, and Mcleod phenotype blood group.
|
|
| Stomatocytes | Stomatocytes are erythrocytes with a loosely folded, mouth-like pale area across the cell. They can be seen in hereditary stomatocytosis, liver disease, Rh null phenotypes, and Tangier's disease.
 |
|
TBack to top ^ | ||
| T-ALL | Also known as T-Lymphoblastic Leukemia/Lymphoma (or T-Acute lymphoblastic leukemia) is an Acute leukemia of lymphoid origin (specifically T-cell origin). Many involve the anterior mediastinum (thymus). Recall the mnemonic of 4 "Ts" for Anterior Mediastinal Lesions: T for Terrible lymphomas (e.g. T-ALL, although Hodgkin lymphoma many times may involve anterior mediastinum and is not as aggressive as T-ALL), T for Thymoma, T for Thyroid remnant, and T for Teratoma (or other germ cell neoplasms). | |
| T-cell | T-cells (AKA T-lymphocyte) are white blood cells involved in cell-mediated immunity along with other cells such as NK cells, etc. T-cell maturation (positive and negative selection) takes place within the Thymus (Remember T for T-cell and T for Thymus). Main subtypes of T-cells include, T-helper cells (CD4+) and cytotoxic T-cells (CD8+).
|
|
| T-cell lymphomas | T-cell lymphomas are a group of Non-Hodgkin lymphomas (NHLs) that are the result of proliferating neoplastic T-lymphocytes (T-cells). They account for approximately 15% of the NHL group. Some of the more common types of T-cell lymphomas include Peripheral T-cell Lymphoma NOS, angioimmunoblastic t-cell lymphoma, Cutaneous T-cell lymphomas (most are Mycoises Fungoides), Anaplastic large cell lymphoma (ALCL), T-LGL Leukemia and Adult T-cell lymphoma/leukemia (ATLL). | |
| Target cells | Target cells are RBCs that resemble a bull's-eye; commonly seen in patients with thalassemia, certain hemoglobinopaties (e.g. Hb C disease), or even liver disease but can also be due to an artifact seen on certain peripheral blood smears secondary to slow drying of blood or excess EDTA.
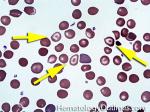  |
|
| Teardrop RBCs | Teardrop cells (dacrocytes) are RBCs that resemble a teardrop in the peripheral blood smear. They are often seen in people with an underlying myelofibrosis or other marrow space occupying disorders.
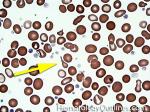  |
|
| Thalassemia | A family of inherited blood disorders that is characterized by decreased production of either alpha or beta-globin chains of the hemoglobin molecule. The two major types of thalassemias are alpha_thalassemia (decreased alpha chain production) and beta_thalassemia (decreased beta chain production). The decreased hemoglobin production results in microcytic (low MCV; <80) red blood cells. Underproduction of one globin chain typically results in stimulating the bone marrow to increase its production of RBCs (as opposed to iron deficiency anemia where there is typically a decrease in RBCs). Hence, in cases of thalassemia the ratio of MCV/RBC is typically lower (<13) than that seen in iron deficiency anemia (usually a ratio of >15). Note: As opposed to hemoglobinopathy which produces abnormal hemoglobin molecules, the hemoglobin molecules produced in thalassemias are decreased but normal in function. |
|
| Thrombopoietin | A protein (hormone) produced by the liver and kidney that stimulates production and differentiation of megakaryocytes and ultimately stimulates the production of more platelets. Note: "Thrombo" refers to platelets and "-poietin" refers to its ability to stimulate production. | |
| Thrombotic Thrombocytopenic Purpura | Also known as TTP is a microangiopathic hemolytic anemia that is typically due to deficiency of ADAMTS-13 (a metalloprotease responsible for cleavage of large multimers of vW factor). Similar to HUS remember the mnemonic "Brain FART" (complements of Dr. M. Baughn) which is "Brain" for the neurologic problems (more commonly associated with TTP than HUS), F for Fever, A for Anemia, R for Renal failure (more commonly associated with HUS than TTP), and T for the Thrombocytopenia. | |
| Thymocyte | An immature T-cell in the thymus. The sequential thymocyte stages can be defined by their location in the thymus (e.g. cortical thymocyte, medullary thymocyte). The stages can also be divided immunophenotypically into CD4/CD8 double negative, CD4/CD8 double positive, and single positive CD4 or CD8. T-cell receptor (TCR) genes rearrangement takes place in the thymus in these thymocytes as they mature into functional CD4+ or CD8+ T-cells. | |
| Tissue Factor | a.k.a. Platelet tissue factor and Factor III. A cell surface protein present on subendothelial cells, platelets, and leukocytes. As a part of extrinsic coagulation pathway, TF contains a receptor which binds Factor VIIa which then converts Factor X to Xa, activating the common coagulation pathway. | |
| Toxic granulation | Toxic granules are dark blue/purple granules seen within the cytoplasm of neutrophils and typically associated with an underlying bacterial infection or sepsis. Toxic granules may be accompanied by toxic vacuoles.
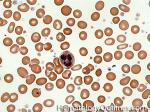  |
|
| Toxic vacuoles | Toxic vacuoles are typically associated with an underlying inflammatory reaction and commonly associated with infections. The cytoplasm of these neutrophils commonly also displays toxic granules and sometimes Dohle_bodies. 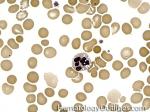 |
|
| TPO | a.k.a. thrombopoietin is a glycoprotein hormone involved in the regulation of platelet production by promoting the proliferation and differentiation of megakaryocytes in the bone marrow. It is produced by the liver and kidney. Currently it is not used as a therapeutic agent. Theoretical uses include platelet count recovery after myelosuppressive chemotherapy, and platelet procurement for donation. | |
| Transferrin | A plasma protein that is produced by the liver and is responsible for iron transport by reversibly binding iron. An increased transferrin level is usually expected in iron deficiency anemia, whereas in iron overload or conditions with excess iron there is usually a decreased level of transferrin. A non-detectable transferrin level could represent atransferrinemia, or familial hypotranferrinemia. | |
| Transformation | Transformation may simply denote enlargement of a cell. But in hematology, transformation is most often used to describe change to a higher grade malignancy (e.g. CLL transformation into DLBCL). | |
| TTP | Stands for Thrombotic Thrombocytopenic Purpura which is a microangiopathic hemolytic anemia (MAHA) that is usually due to a deficiency of ADAMTS-13 (a metalloprotease responsible for cleavage of large multimers of vW factor). Similar to hemolytic uremic syndrome (HUS) remember the mnemonic "Brain FART" (Complements of Dr. M. Baughn) which is Brain for neurologic problems (more commonly associated with TTP than HUS), F for Fever, A for Anemia, R for Renal failure (more commonly associated with HUS than TTP), and T for Thrombocytopenia. On peripheral smear, usually schistocytes are easily notable and increased in number (similar to other MAHA). |
|
UBack to top ^ | ||
| U-- | - | |
VBack to top ^ | ||
| Vacuolated cells | Cells that contain vacuoles within the cytoplasm. The vacuoles are typically thought to be of lysosomal origin as seen in mature circulating monocytes in blood. Cells that typically have vacuoles include but are not limited to neutrophils, monocytes, monoblasts, abnormal erythroid precursors (e.g. Erythroid Leukemia), and Burkitt Lymphoma's leukemic phase. | |
| Vitamin B12 | Vitamin B12 is a.k.a cobalamine is a water-soluble vitamin involved in CNS functioning and formation of red blood cells. Vitamin B12 deficiency is usually associated with megaloblastic macrocytic anemia due to defective DNA synthesis, and may ultimately lead to neurologic damage. Intrinsic factor (IF), produced by the stomach, is crucial for vitamin B12 absorption, and lack of IF can lead to B12 deficiency and pernicious anemia. Vitamin B12 deficiency results in elevated serum levels of homocysteine and methylmalonic acid. First, vitamin B12 is use in the generation of tetrahydrofolate, which is used in the conversion of homocysteine to methionine. Second, B12 is a cofactor in the conversion of methylmalonyl-CoA to succinyl-CoA in the Kreb cycle. Therefore, laboratory tests for homocysteine or methylmalonic acid levels can be used as indicators for B12 deficiency as well in conjunction with B12 levels. Take home points: B12 deficiency may show both hyperhomocyteinemia and an elevated MMA (Methyl Malonic Acid) while folate deficiency may show hyperhomocyteinemia but no elevation in MMA. |
|
| Vitamin K | Fat-soluble vitamin required in coagulation and metabolic pathways. The Vitamin K dependent Coagulation factors are II, VII, IX, X, Protein C & S. | |
| von Willebrand Disease | A hereditary or acquired coagulation disorder resulting from either a qualitative change of vWF (usually a mutation giving rise to an abnormal vWF) or quantitative deficiency of vWF. There are three types of hereditary vWF deficiencies: Type 1, Type 2, and Type 3. Type 1 is the most common form and is a quantitative defect where vWF are decreased. Most patients are asymptomatic; however, symtoms such as prolonged bleeding post-surgery, menorrhagia, or easy bruising can occur. Type 2 is subdivided into 2A, 2B, 2M, and 2N, and it is a qualitative defect of vWF. Type 2B is unique in that the mutation leads to an increased binding of platelets to vWF and increased clearance thus producing a mild thrombocytopenia. Type 2M and 2N are rare. Type 3 is the most severe form and has no detectable levels of vWF. | |
| von Willebrand Factor | a.k.a. vWF is a multimeric protein produced in the endothelium and subendothelial tissues, and also in the alpha granules within platelets. vWF is bound to Factor VIII while inactive and released by the influence of thrombin and binds to subendothelial collagen during vessel trauma and damage. vWF then binds to platelets via the GPIb receptor. Additional platelets bind via vWF and the process repeats. The breakdown of vWF multimers is by ADAMTS13, a metalloproteinase. | |
| vWF | a.k.a. von willebrand factor is a multimeric protein produced in the endothelium and subendothelial tissues, and also in the alpha granules within platelets. vWF is bound to Factor VIII while inactive and released by the influence of thrombin and binds to subendothelial collagen during vessel trauma and damage. vWF then binds to platelets via the GPIb receptor. Additional platelets bind via vWF and the process repeats. The breakdown of vWF multimers is by ADAMTS13, a metalloproteinase. | |
WBack to top ^ | ||
| Waldenstrom's macroglobulinemia | Waldenstrom's macroglobulinemia (WM) is a clinical disease usually caused by an increase in abnormal B-lymphocytes which produce increased amounts of monoclonal immunoglobulins (typically IgM). Very high levels of IgM in the blood may cause hyperviscosity , which could explain the symptoms associated with this disease (blurred vision, headache, and dizziness). The Lymphoma that is most commonly associated with WM is lymphoplasmacytic lymphoma (a low grade B-cell lymphoma). | |
| WBC | White blood Cells are the nucleated cells noted in the peripheral blood as part of a CBC and include neutrophils, lymphocytes, monocytes, eosinophils, and basophils (in order of prevalence). | |
| White Blood Cells | Also known as WBC which are the nucleated cells noted in the peripheral blood as part of a CBC and include neutrophils, lymphocytes, monocytes, eosinophils, and basophils (in order of prevalence). | |
XBack to top ^ | ||
| X-- | - | |
YBack to top ^ | ||
| Y-- | - | |
ZBack to top ^ | ||
| Zeta potential | The electrical potential difference between red blood cells created by the surface negative charges on the red blood cells and the surrounding cations in the blood. This repulsive force prevents the RBCs from sticking together under normal conditions. Conditions that decrease this repulsion are typically relatively positively charged molecules which increases the chance of RBC agglutination (e.g. increased Ig levels as in plasma cell Myeloma or changing the ionic strength when adding Low Ionic Strength Solution (LISS) in certain blood bank tests, etc.) |
|